GTEx V6
Yuxin Zou
2018-12-20
Last updated: 2019-01-01
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(20181220)The command
set.seed(20181220)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 26398fd
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .DS_Store Ignored: .Rhistory Ignored: .Rproj.user/ Ignored: analysis/figure/ Ignored: output/.DS_Store Untracked files: Untracked: code/ED.R Untracked: code/mCurrent.R Untracked: code/mIgnore.R Untracked: code/mMLE.R Untracked: code/mSimple.R Untracked: code/mV3.R Untracked: data/cor_tissues_non_ash_voom_pearson.rda Untracked: data/gene_names_GTEX_V6.txt Untracked: data/genewide_ash_out_tissue_mat_halfuniform_non_mode.rda Untracked: data/order_index.rda Untracked: data/samples_id.txt Untracked: data/tissuewide_pearson_halfuniform_tissuewide_non_mode.rda Untracked: output/GTExV6/
Expand here to see past versions:
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 26398fd | zouyuxin | 2019-01-01 | wflow_publish(“analysis/GTExV6.Rmd”) |
library(flashr)
library(mixsqp)
library(mashr)Loading required package: ashrlibrary(mclust)Package 'mclust' version 5.4.1
Type 'citation("mclust")' for citing this R package in publications.
Attaching package: 'mclust'The following object is masked from 'package:ashr':
denslibrary(knitr)
library(kableExtra)
library(ggplot2)gtex <- readRDS(gzcon(url("https://github.com/stephenslab/gtexresults/blob/master/data/MatrixEQTLSumStats.Portable.Z.rds?raw=TRUE")))strong.z = gtex$strong.z
data.strong = mash_set_data(strong.z)
data.random = mash_set_data(gtex$random.b, gtex$random.s)Data Driven Covariances
Flash:
my_init_fn <- function(Y, K = 1) {
ret = flashr:::udv_si(Y, K)
pos_sum = sum(ret$v[ret$v > 0])
neg_sum = -sum(ret$v[ret$v < 0])
if (neg_sum > pos_sum) {
return(list(u = -ret$u, d = ret$d, v = -ret$v))
} else
return(ret)
}
flash_pipeline = function(data, ...) {
## current state-of-the art
## suggested by Jason Willwerscheid
## cf: discussion section of
## https://willwerscheid.github.io/MASHvFLASH/MASHvFLASHnn2.html
ebnm_fn = "ebnm_ash"
ebnm_param = list(l = list(mixcompdist = "normal",
optmethod = "mixSQP"),
f = list(mixcompdist = "+uniform",
optmethod = "mixSQP"))
##
fl_g <- flashr:::flash_greedy_workhorse(data,
var_type = "constant",
ebnm_fn = ebnm_fn,
ebnm_param = ebnm_param,
init_fn = "my_init_fn",
stopping_rule = "factors",
tol = 1e-3,
verbose_output = "odF")
fl_b <- flashr:::flash_backfit_workhorse(data,
f_init = fl_g,
var_type = "constant",
ebnm_fn = ebnm_fn,
ebnm_param = ebnm_param,
stopping_rule = "factors",
tol = 1e-3,
verbose_output = "odF")
return(fl_b)
}
cov_flash = function(data, subset = NULL, non_canonical = FALSE, save_model = NULL) {
if(is.null(subset)) subset = 1:mashr:::n_effects(data)
b.center = apply(data$Bhat, 2, function(x) x - mean(x))
## Only keep factors with at least two values greater than 1 / sqrt(n)
find_nonunique_effects <- function(fl) {
thresh <- 1/sqrt(ncol(fl$fitted_values))
vals_above_avg <- colSums(fl$ldf$f > thresh)
nonuniq_effects <- which(vals_above_avg > 1)
return(fl$ldf$f[, nonuniq_effects, drop = FALSE])
}
fmodel = flash_pipeline(b.center)
if (non_canonical)
flash_f = find_nonunique_effects(fmodel)
else
flash_f = fmodel$ldf$f
## row.names(flash_f) = colnames(b)
if (!is.null(save_model)) saveRDS(list(model=fmodel, factors=flash_f), save_model)
if(ncol(flash_f) == 0){
U.flash = list("tFLASH" = t(fmodel$fitted_values) %*% fmodel$fitted_values / nrow(fmodel$fitted_values))
} else{
U.flash = c(cov_from_factors(t(as.matrix(flash_f)), "FLASH"),
list("tFLASH" = t(fmodel$fitted_values) %*% fmodel$fitted_values / nrow(fmodel$fitted_values)))
}
return(U.flash)
}U.f = cov_flash(data.strong, non_canonical = TRUE, save_model = '../output/GTExV6/flash_model.rds')
saveRDS(U.f, 'output/GTExV6/flash_cov.rds')missing.tissues <- c(7, 8, 19, 20, 24, 25, 31, 34, 37)
gtex.colors <- read.table("https://github.com/stephenslab/gtexresults/blob/master/data/GTExColors.txt?raw=TRUE", sep = '\t', comment.char = '')[-missing.tissues, 2]
gtex.colors <- as.character(gtex.colors)
fl_model = readRDS('output/GTExV6/flash_model.rds')$model
factors = readRDS('output/GTExV6/flash_model.rds')$factors
par(mfrow = c(2, 3))
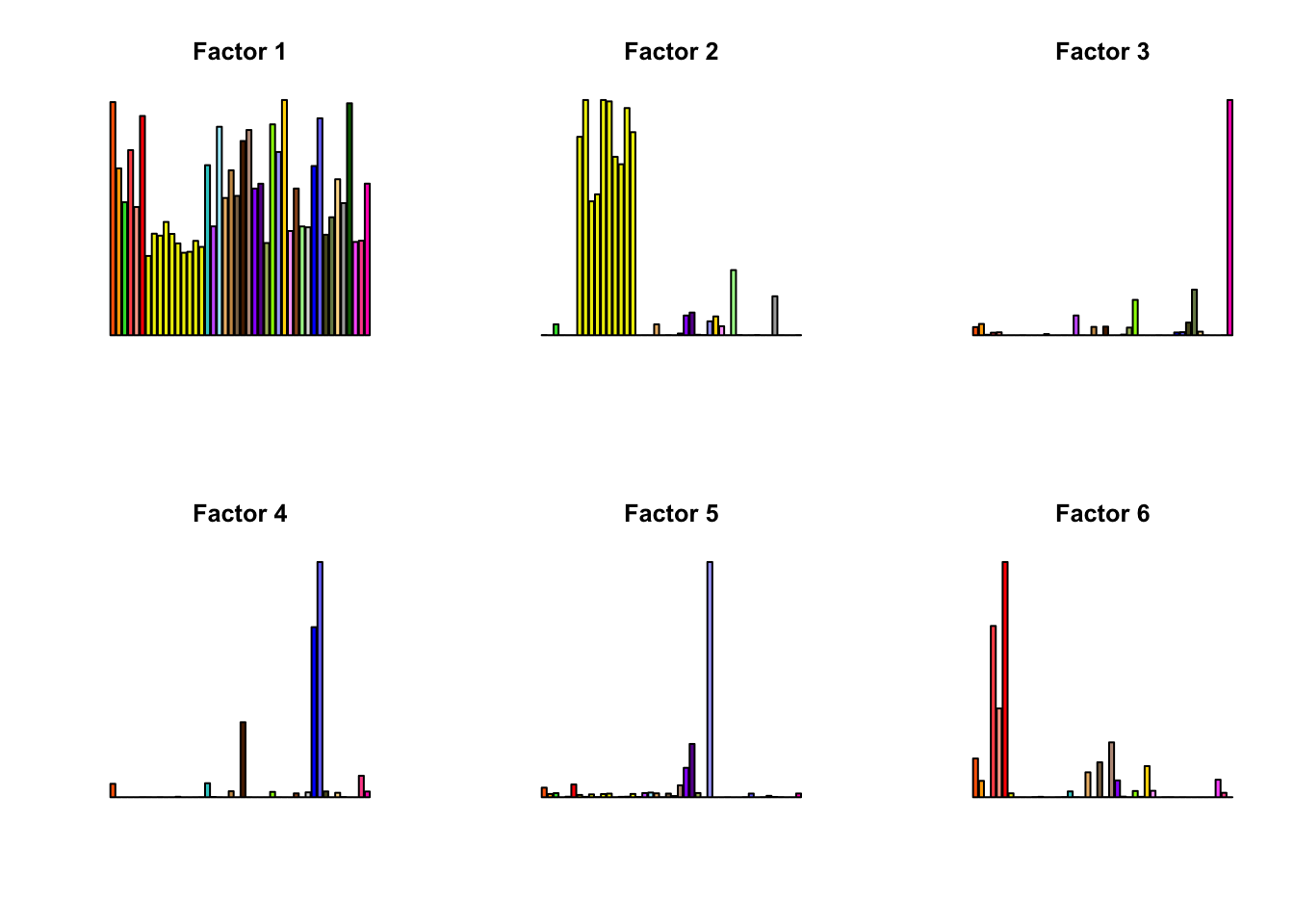
for(k in 1:16){
barplot(factors[,k], col=gtex.colors, names.arg = FALSE, axes = FALSE, main=paste0("Factor ", k))
}
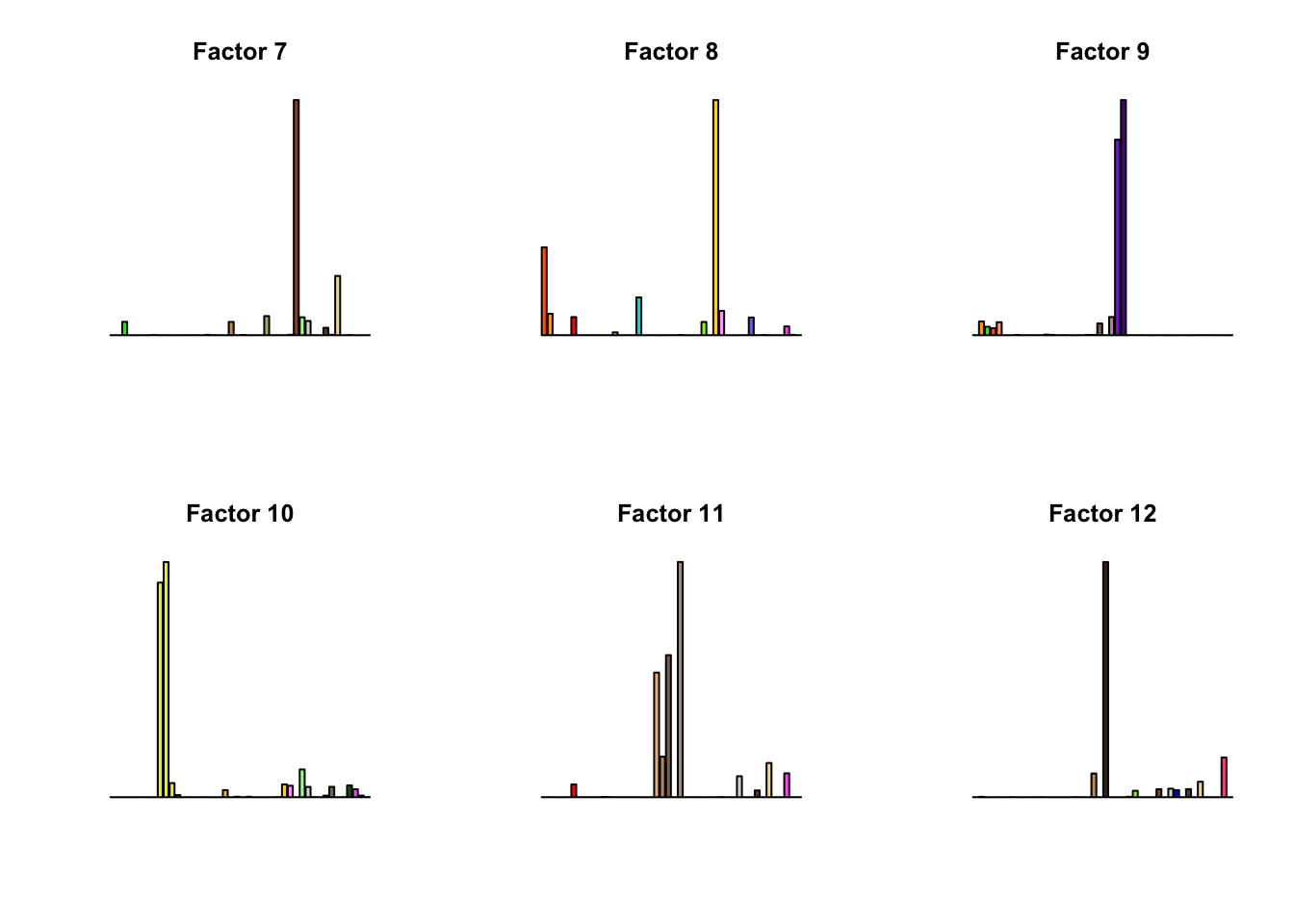
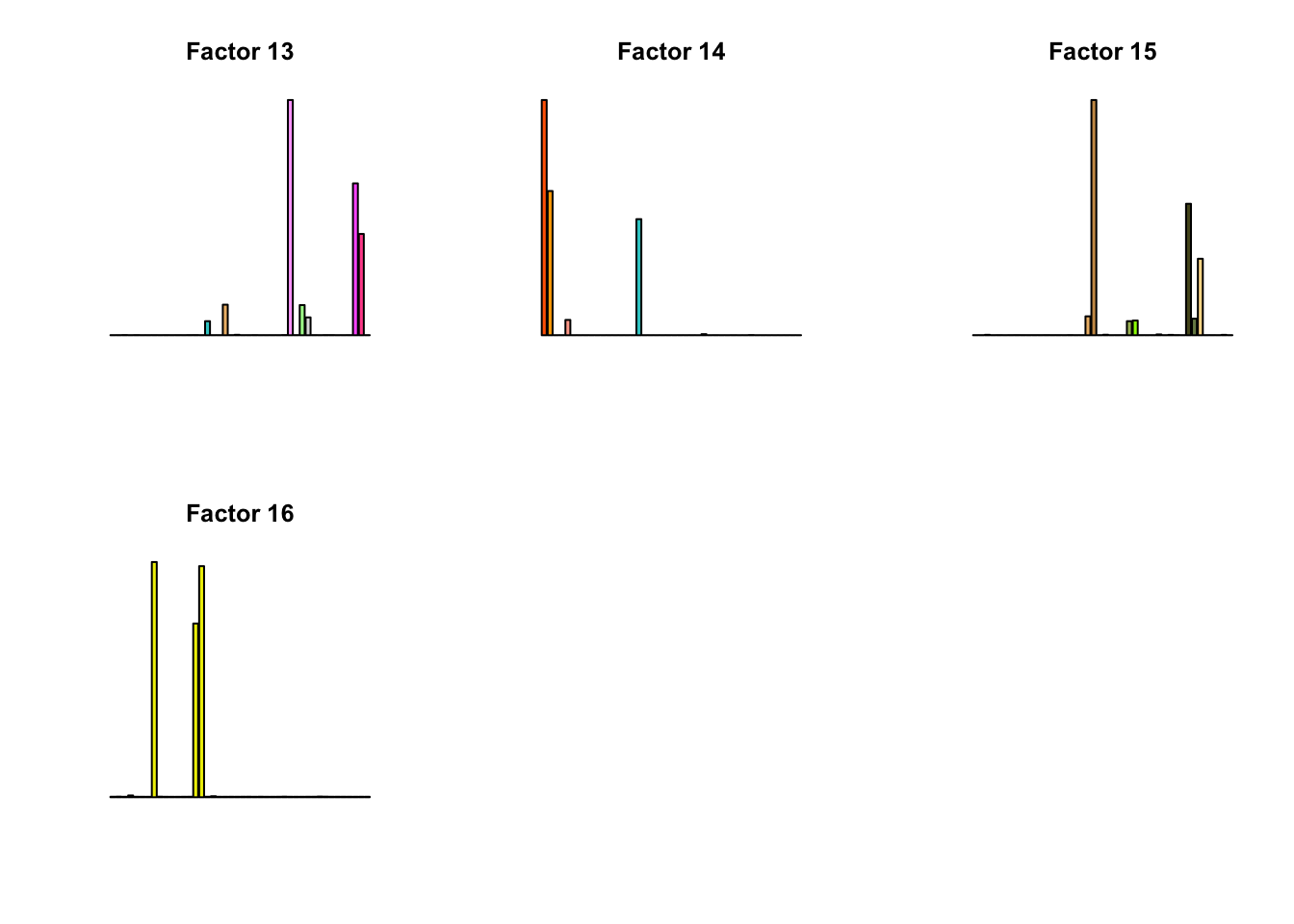
fll_model = flash_pipeline(fl_model$ldf$l)
saveRDS(fll_model, 'output/GTExV6/flash_loading_model.rds')U.pca = cov_pca(data.strong, 5)U.ed = cov_ed(data.strong, c(U.f, U.pca))U.ed = readRDS('output/GTExV6/Ued.rds')U.c = cov_canonical(data.random)data.strong = mash_set_data(Bhat = gtex$strong.b, Shat = gtex$strong.s)m.ignore = mash(data.random, c(U.c, U.ed), outputlevel = 1)
m.ignore$result = mash_compute_posterior_matrices(m.ignore, data.strong)V.simple = estimate_null_correlation_simple(data.random)data.random.V.simple = mash_update_data(data.random, V = V.simple)
m.simple = mash(data.random.V.simple, c(U.c, U.ed), outputlevel = 1)
data.strong.V.simple = mash_update_data(data.strong, V = V.simple)
m.simple$result = mash_compute_posterior_matrices(m.simple, data.strong.V.simple)set.seed(1)
random.subset = sample(1:nrow(gtex$random.b),5000)
data.random.s = mash_set_data(gtex$random.b[random.subset,], gtex$random.s[random.subset,])
current = estimate_null_correlation(data.random.s, c(U.c, U.ed), max_iter = 6)
V.current = current$V
data.random.V.current = mash_update_data(data.random, V = V.current)
m.current = mash(data.random.V.current, c(U.c, U.ed), outputlevel = 1)
data.strong = mash_update_data(data.strong, V = V.current)
m.current$result = mash_compute_posterior_matrices(m.current, data.strong)V = get(load('~/Documents/GitHub/GTEx/data/genewide_ash_out_tissue_mat_halfuniform_non_mode.rda'))
# select tissue
tissue_labels <- read.table(file = "~/Documents/GitHub/GTEx/data/samples_id.txt")[,3]
U <- unique(tissue_labels)
tissues = c(1:6, 9:18, 21:23,26:30,32:33,35:36,38:53)
V = V[tissues,tissues,]
V.strong = V
for(i in 1:nrow(gtex$strong.b)){
V.strong[,,i] = as.matrix(Matrix::nearPD(V[,,i], conv.tol=1.e-05, corr = TRUE, maxit = 200, doSym = TRUE)$mat)
}
saveRDS(V.strong, 'output/GTExV6/V_strong_genewide.rds')
# select genes
gene_names <- as.character(read.table(file = "~/Documents/GitHub/GTEx/data/gene_names_GTEX_V6.txt")[,1])
gene_names_1 <- as.character(sapply(gene_names, function(x) return(strsplit(x, "[.]")[[1]][1])))
data.random.names = rownames(data.random$Bhat)
data.random.names.1 = as.character(sapply(data.random.names, function(x) return(strsplit(x, "[.]")[[1]][1])))
V.random = array(NA, dim = c(44,44,nrow(gtex$random.b)))
for(i in 1:nrow(gtex$random.b)){
numg <- grep(data.random.names.1[i], gene_names_1)
V.random[,,i] = as.matrix(Matrix::nearPD(V[,,numg], conv.tol=1.e-05, corr = TRUE, doSym = TRUE)$mat)
}
saveRDS(V.random, 'output/GTExV6/V_random_genewide.rds')
data.random.V3 = mash_update_data(data.random, V = V.random)
m.V3 = mash(data.random.V3, c(U.c, U.ed), outputlevel = 1, algorithm.version = 'R')
data.strong.V3 = mash_update_data(data.strong, V = V.strong)
m.V3$result = mash_compute_posterior_matrices(m.V3, data.strong.V3, algorithm.version = 'R')# read model
m_ignore = readRDS('output/GTExV6/m_ignore_post.rds')
m_ignore_EZ = readRDS('output/GTExV6/m_ignore_EZ_post.rds')
m_simple = readRDS('output/GTExV6/m_simple_post.rds')
m_simple_EZ = readRDS('output/GTExV6/m_simple_EZ_post.rds')
m_current = readRDS('output/GTExV6/m_current_post.rds')
m_current_EZ = readRDS('output/GTExV6/m_current_EZ_post.rds')
m_V3 = readRDS('output/GTExV6/m_V3_genewide_post.rds')
m_V3_EZ = readRDS('output/GTExV6/m_V3_genewide_EZ_post.rds')par(mfrow = c(1,2))
# pdf('../output/GTExV6/Figures/SimpleV.pdf')
corrplot::corrplot(V.simple, method='color', type='upper', tl.col="black", tl.srt=45, tl.cex = 0.7, diag = FALSE, col=colorRampPalette(c("blue", "white", "red"))(200), cl.lim = c(-1,1), title = 'Simple', mar=c(0,0,5,0))Warning in corrplot::corrplot(V.simple, method = "color", type = "upper", :
Not been able to calculate text margin, please try again with a clean new
empty window using {plot.new(); dev.off()} or reduce tl.cex# dev.off()
V.current = readRDS('output/GTExV6/currentV_EZ.rds')
V.current = V.current$V
# pdf('../output/GTExV6/Figures/CurrentEZV.pdf')
corrplot::corrplot(V.current, method='color', type='upper', tl.col="black", tl.srt=45, tl.cex = 0.7, diag = FALSE, col=colorRampPalette(c("blue", "white", "red"))(200), cl.lim = c(-1,1), title = 'Current EZ', mar=c(0,0,5,0))Warning in corrplot::corrplot(V.current, method = "color", type =
"upper", : Not been able to calculate text margin, please try again with a
clean new empty window using {plot.new(); dev.off()} or reduce tl.cex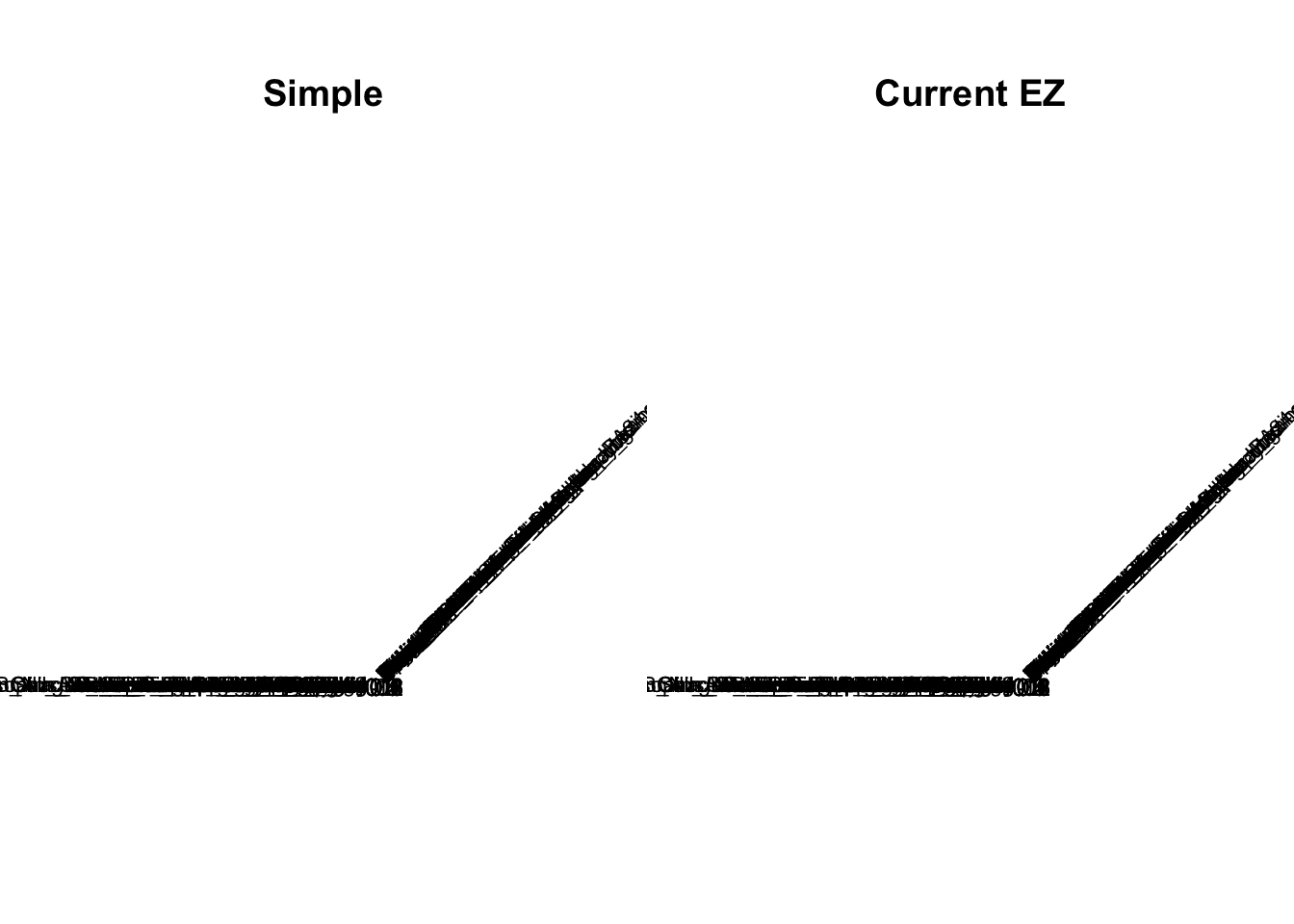
# dev.off()logliks = c(get_loglik(m_ignore), get_loglik(m_simple), get_loglik(m_current), get_loglik(m_V3))
logliks_EZ = c(get_loglik(m_ignore_EZ), get_loglik(m_simple_EZ), get_loglik(m_current_EZ), get_loglik(m_V3_EZ))
tmp = cbind(logliks, logliks_EZ)
row.names(tmp) = c('Ignore', 'Simple', 'Current', 'V3')
colnames(tmp) = c('EE', 'EZ')
tmp %>% kable() %>% kable_styling()| EE | EZ | |
|---|---|---|
| Ignore | 929188.2 | 935288.4 |
| Simple | 933359.2 | 936783.2 |
| Current | 937323.7 | 939685.2 |
| V3 | 860927.1 | 897029.6 |
barplot(get_estimated_pi(m_ignore), las=2, cex.names = 0.7, main = 'Ignore EE')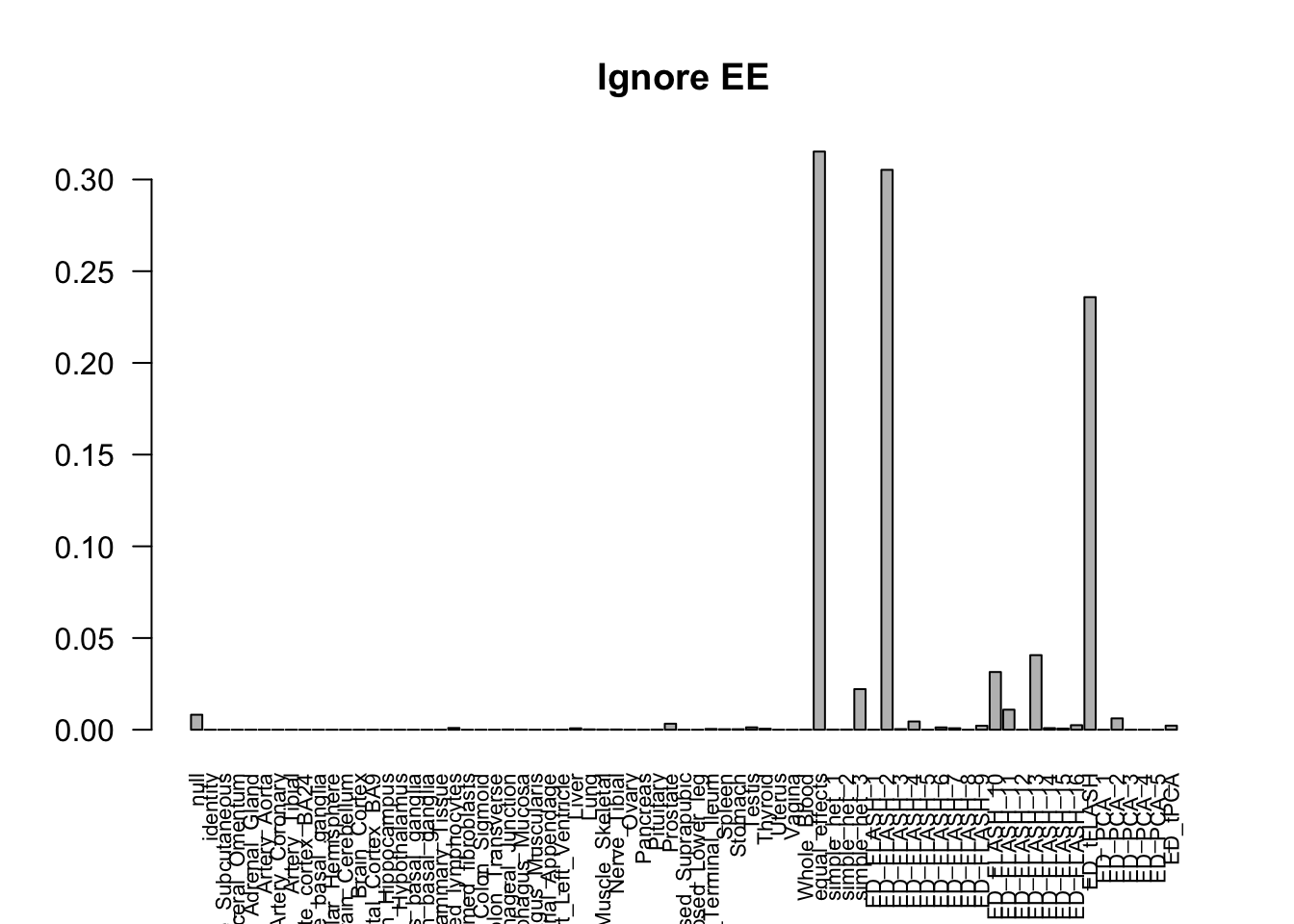
barplot(get_estimated_pi(m_simple), las=2, cex.names = 0.7, main = 'Simple EE')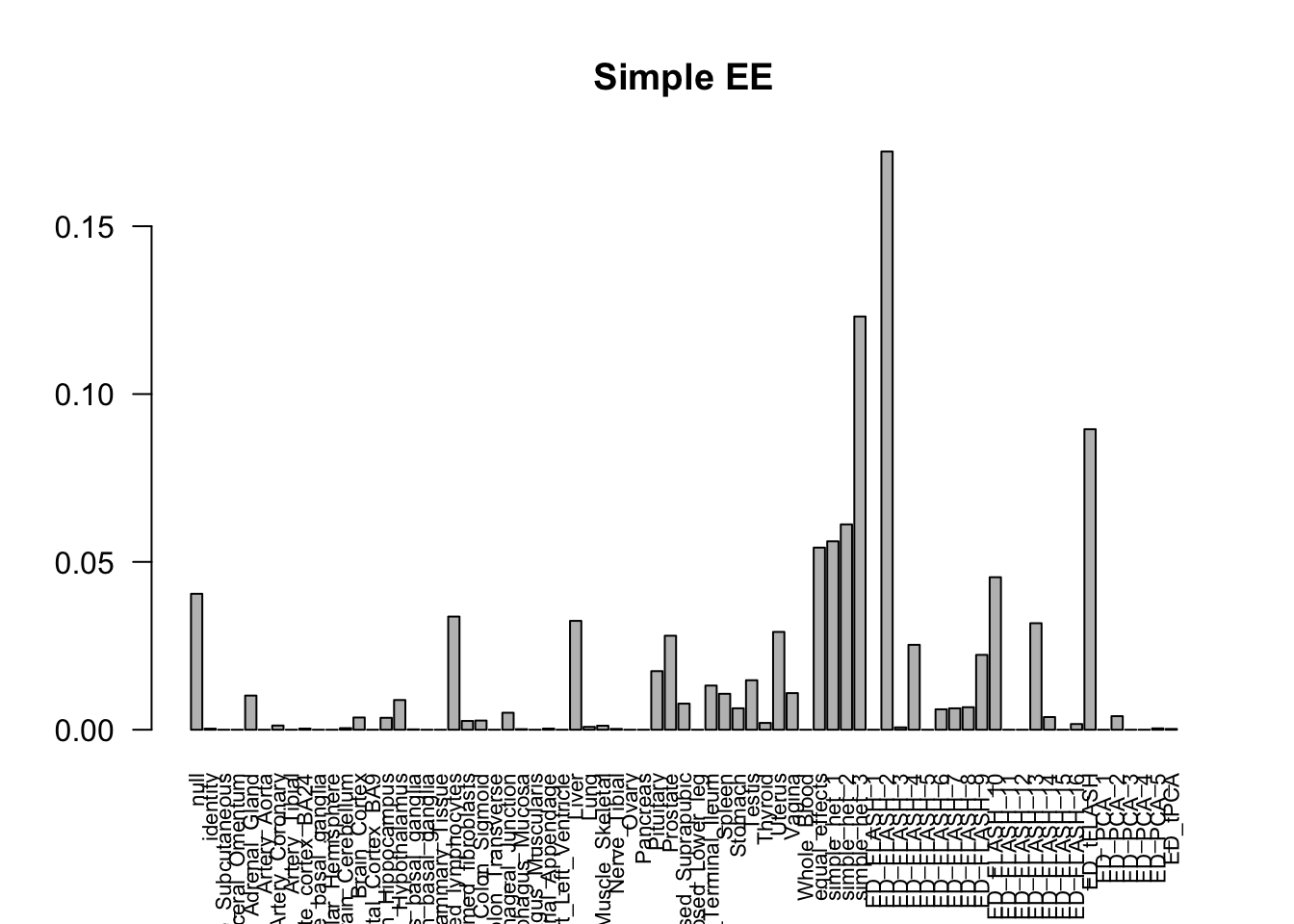
barplot(get_estimated_pi(m_current), las=2, cex.names = 0.7, main = 'Current EE')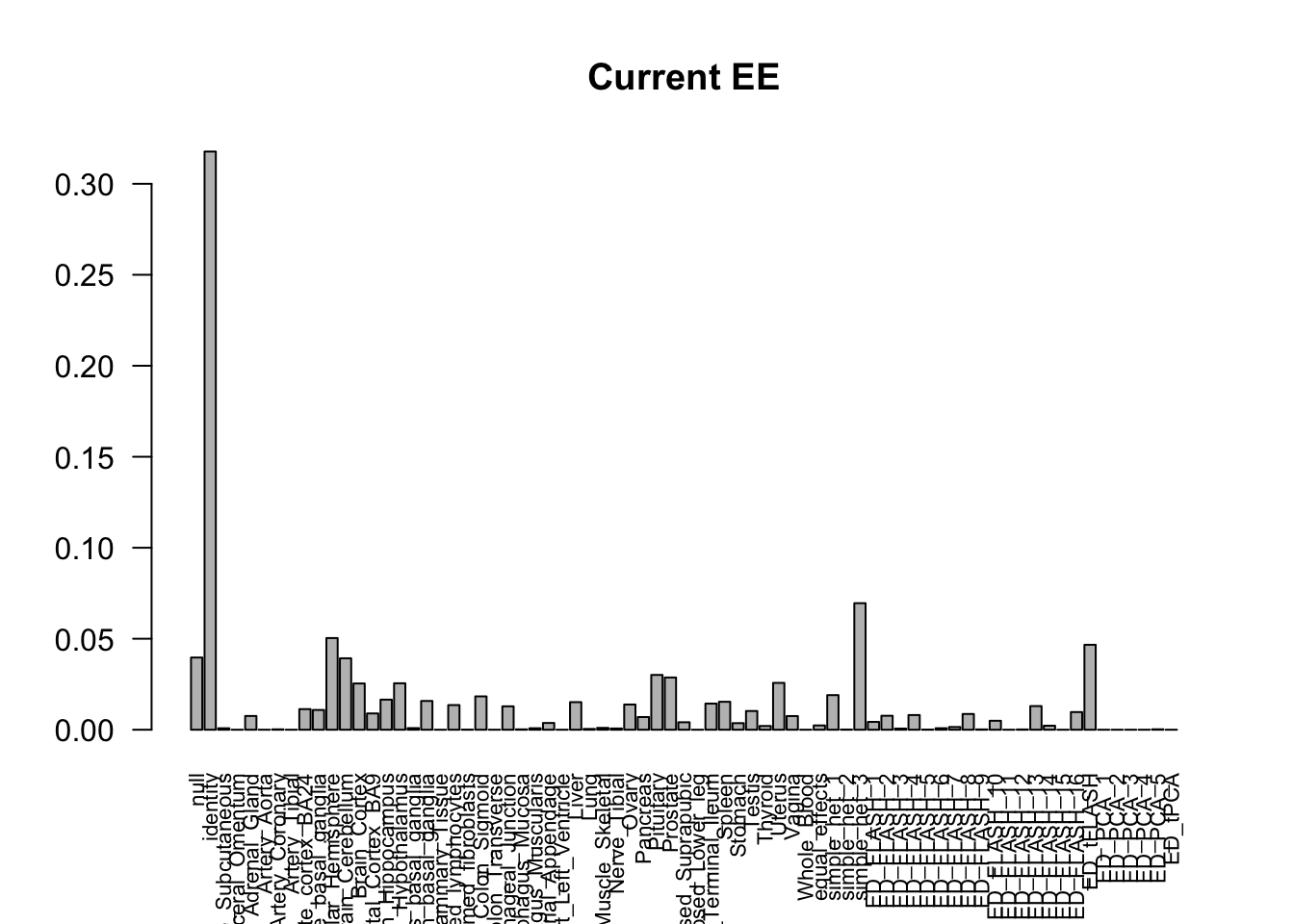
barplot(get_estimated_pi(m_V3), las=2, cex.names = 0.7, main = 'V3 EE')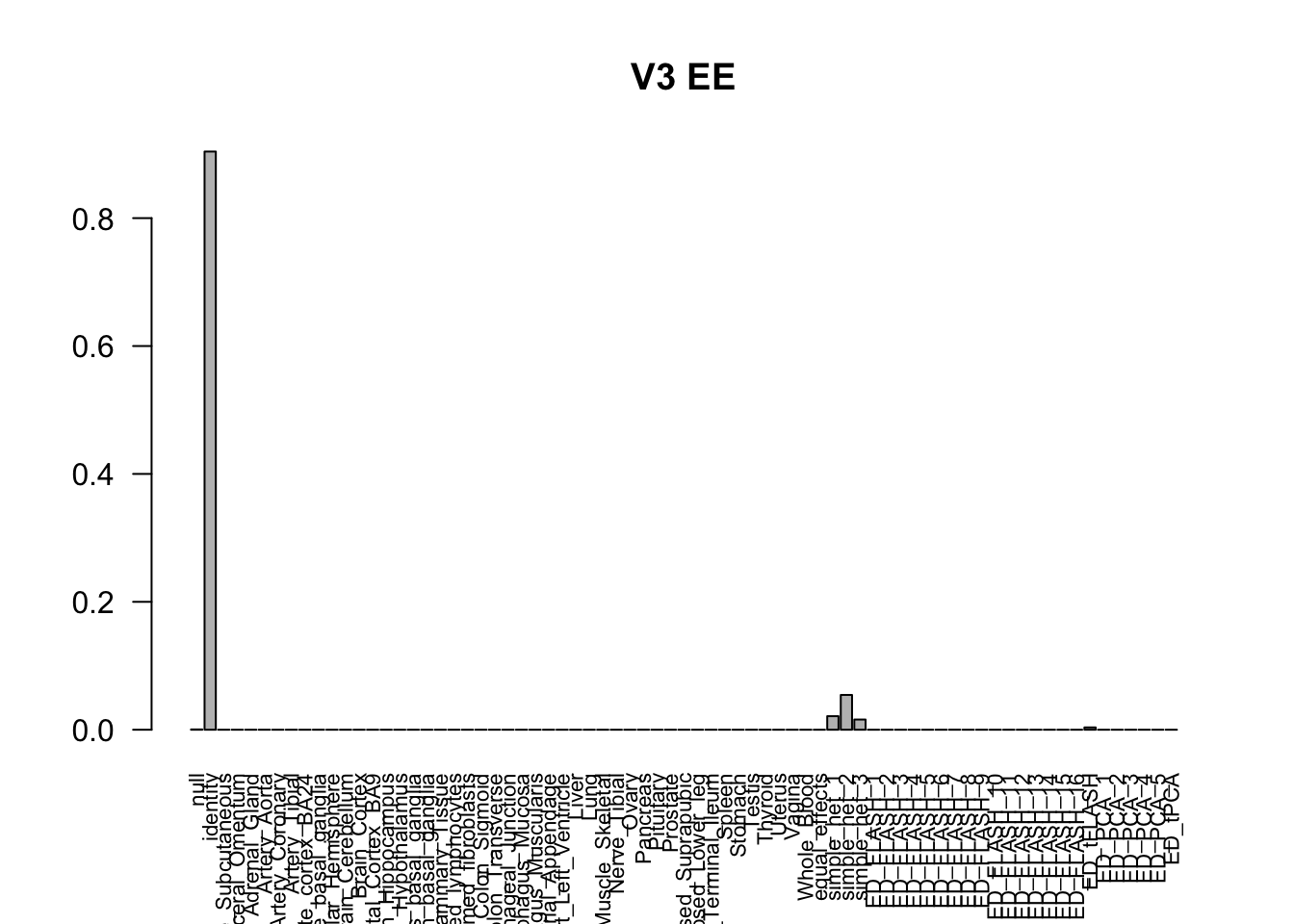
barplot(get_estimated_pi(m_ignore_EZ), las=2, cex.names = 0.7, main = 'Ignore EZ')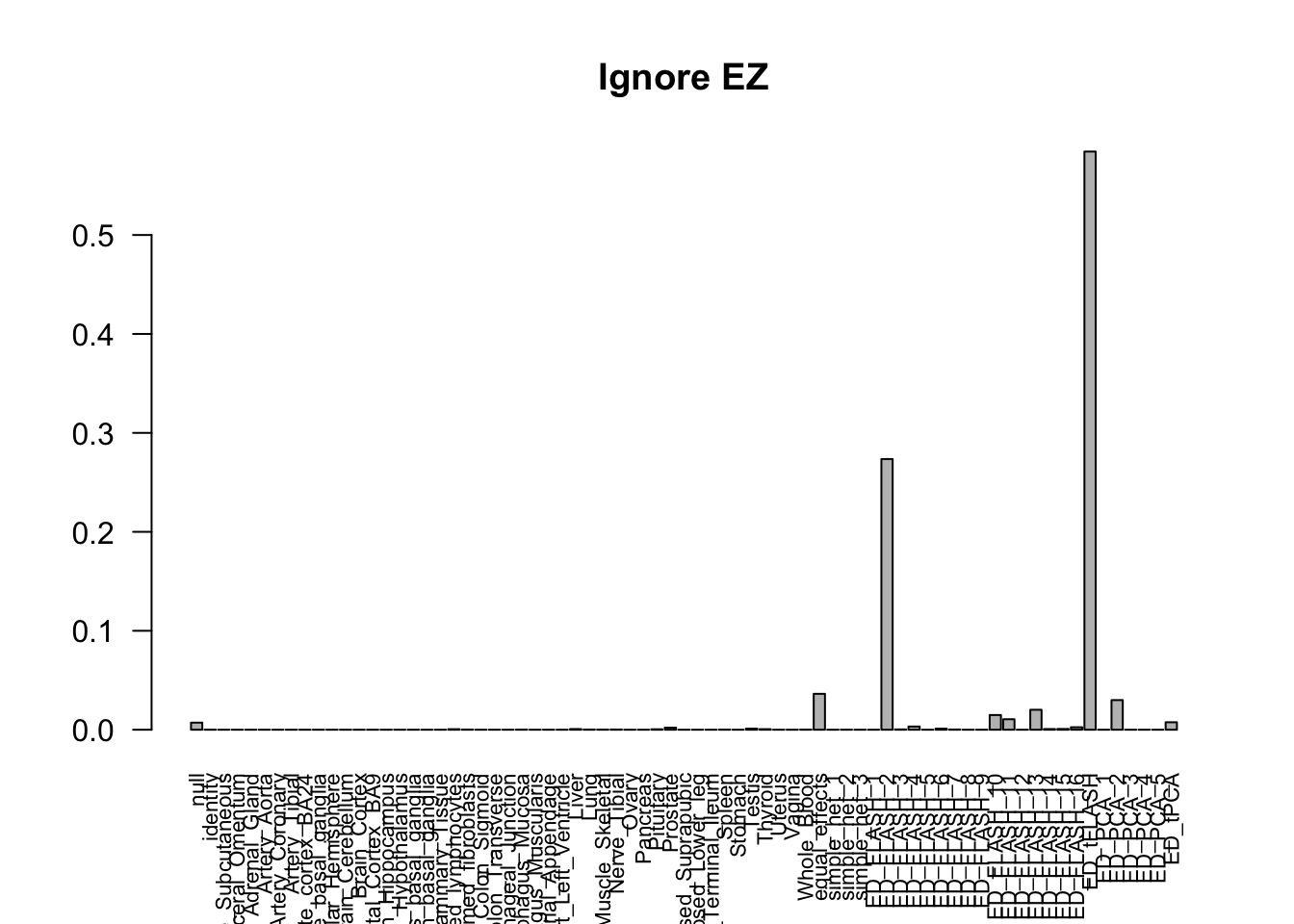
barplot(get_estimated_pi(m_simple_EZ), las=2, cex.names = 0.7, main = 'Simple EZ')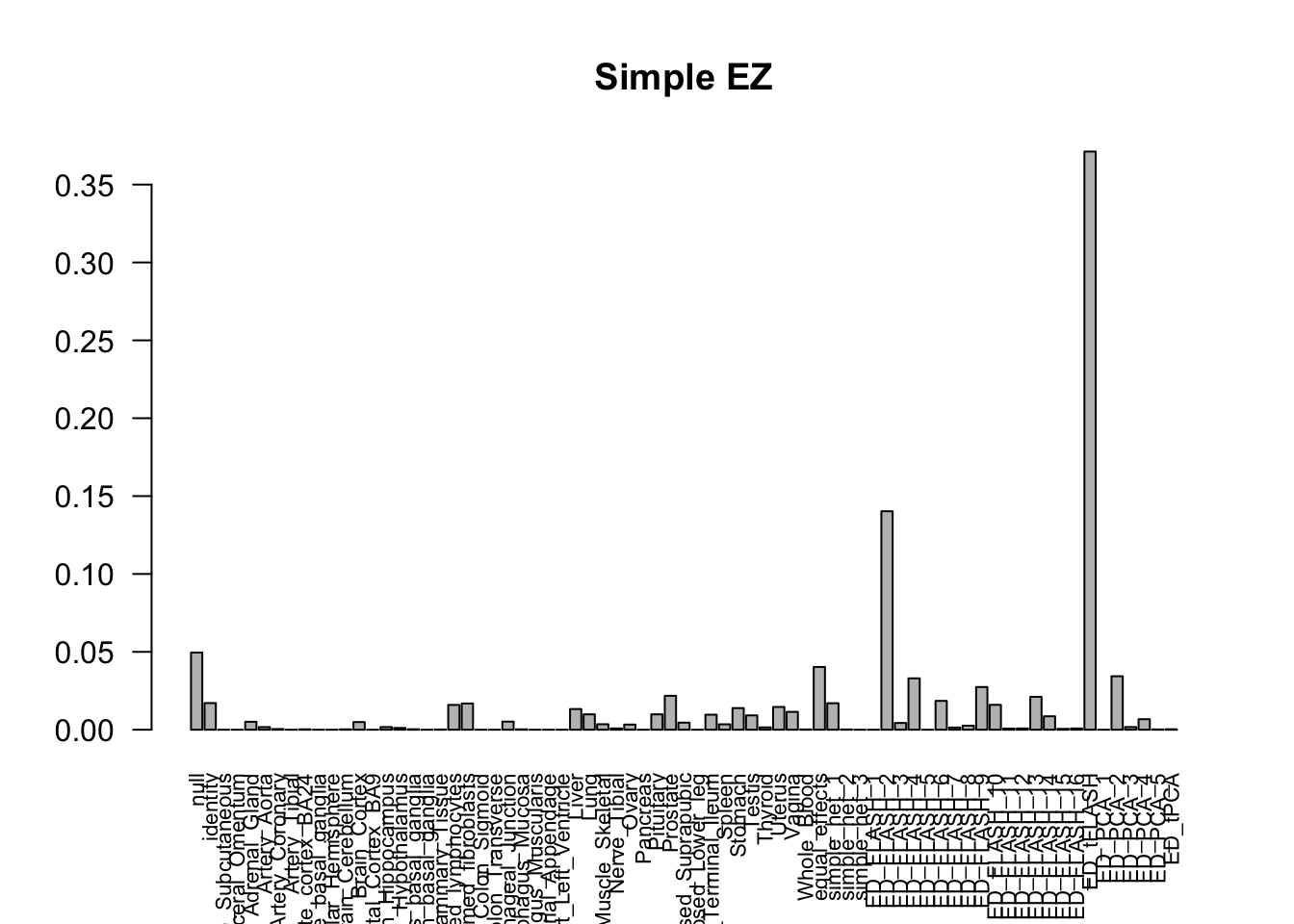
barplot(get_estimated_pi(m_current_EZ), las=2, cex.names = 0.7, main = 'Current EZ')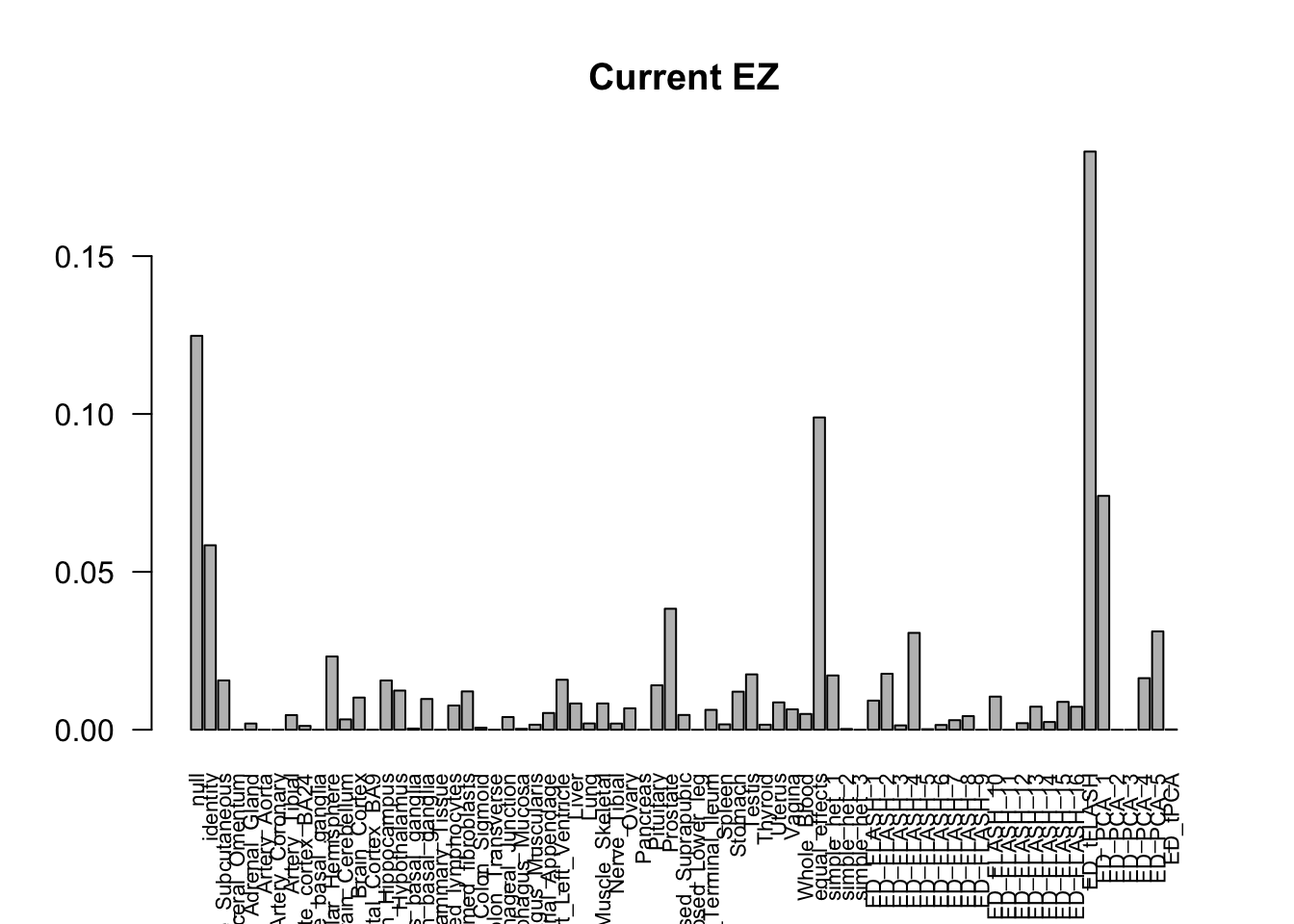
barplot(get_estimated_pi(m_V3_EZ), las=2, cex.names = 0.7, main = 'V3 EZ')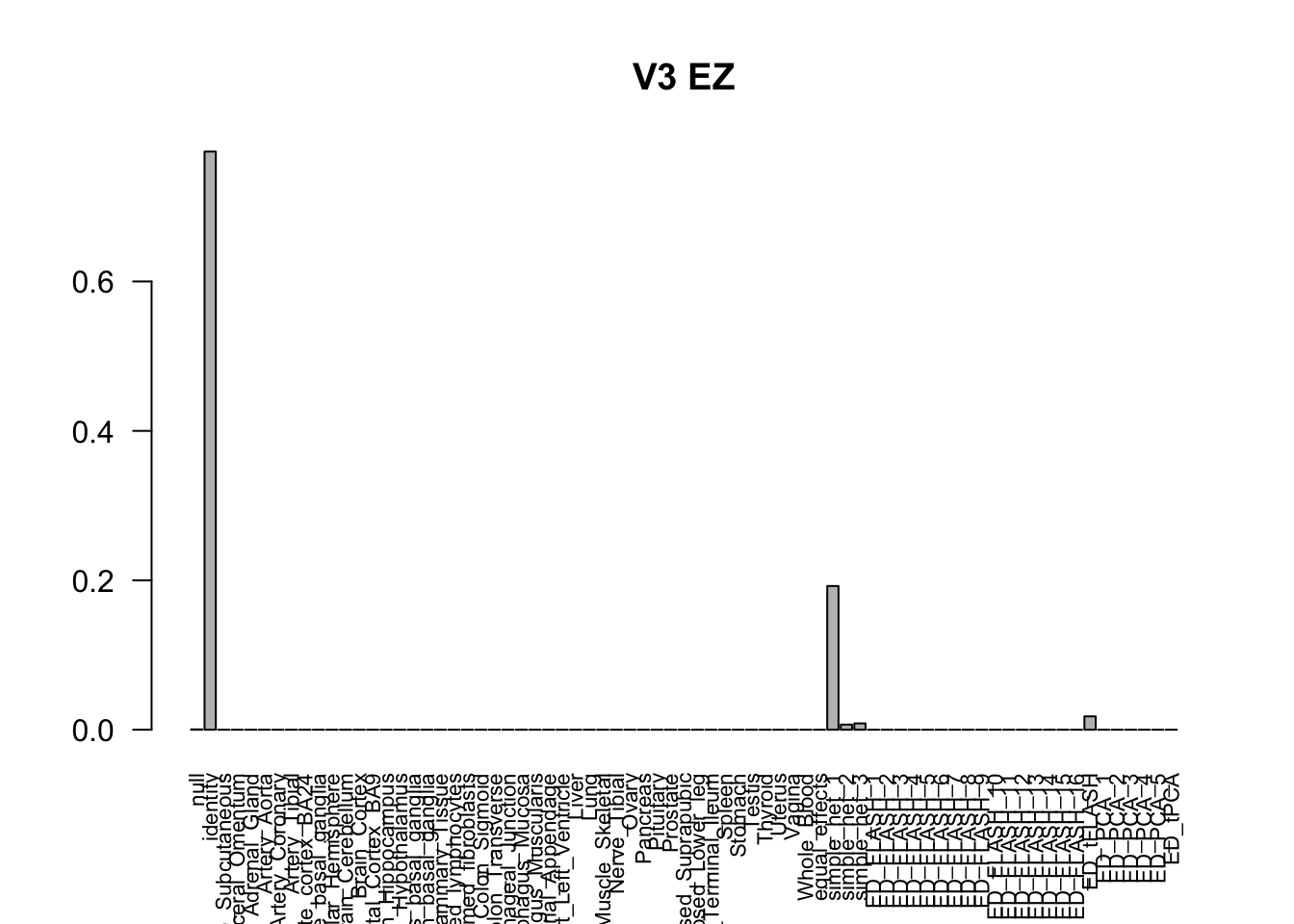
Number of significant:
numsig_EE = c(length(get_significant_results(m_ignore)),
length(get_significant_results(m_simple)),
length(get_significant_results(m_current)),
length(get_significant_results(m_V3)))
numsig_EZ = c(length(get_significant_results(m_ignore_EZ)),
length(get_significant_results(m_simple_EZ)),
length(get_significant_results(m_current_EZ)),
length(get_significant_results(m_V3_EZ)))
tmp = cbind(numsig_EE, numsig_EZ)
row.names(tmp) = c('Ignore', 'Simple', 'Current', 'V3')
colnames(tmp) = c('EE', 'EZ')
tmp %>% kable() %>% kable_styling()| EE | EZ | |
|---|---|---|
| Ignore | 14017 | 14221 |
| Simple | 13037 | 13485 |
| Current | 12803 | 13006 |
| V3 | 16054 | 16069 |
The V3 model has more significant findings, which includes all significant findings in the other three models.
length(intersect(get_significant_results(m_V3_EZ), get_significant_results(m_current_EZ)))[1] 13006length(intersect(get_significant_results(m_V3_EZ), get_significant_results(m_simple_EZ)))[1] 13485length(intersect(get_significant_results(m_V3_EZ), get_significant_results(m_ignore_EZ)))[1] 14221The gene significant in simple EZ, not in current EZ:
par(mfrow=c(2,2))
stronggene = data.frame(gtex$strong.b[5034,])
colnames(stronggene) = 'EffectSize'
stronggene$Group = row.names(stronggene)
stronggene$se = gtex$strong.s[5034,]
ggplot(stronggene, aes(y = EffectSize, x = Group)) +
geom_point(show.legend = FALSE, color=gtex.colors) + coord_flip() + ggtitle('ENSG00000135315') + ylim(c(-0.7,1)) + geom_errorbar(aes(ymin=EffectSize-1.96*se, ymax=EffectSize+1.96*se), width=0.4, show.legend = FALSE, color=gtex.colors) +
theme_bw(base_size=12) + theme(axis.text.y = element_text(colour = gtex.colors, size = 6))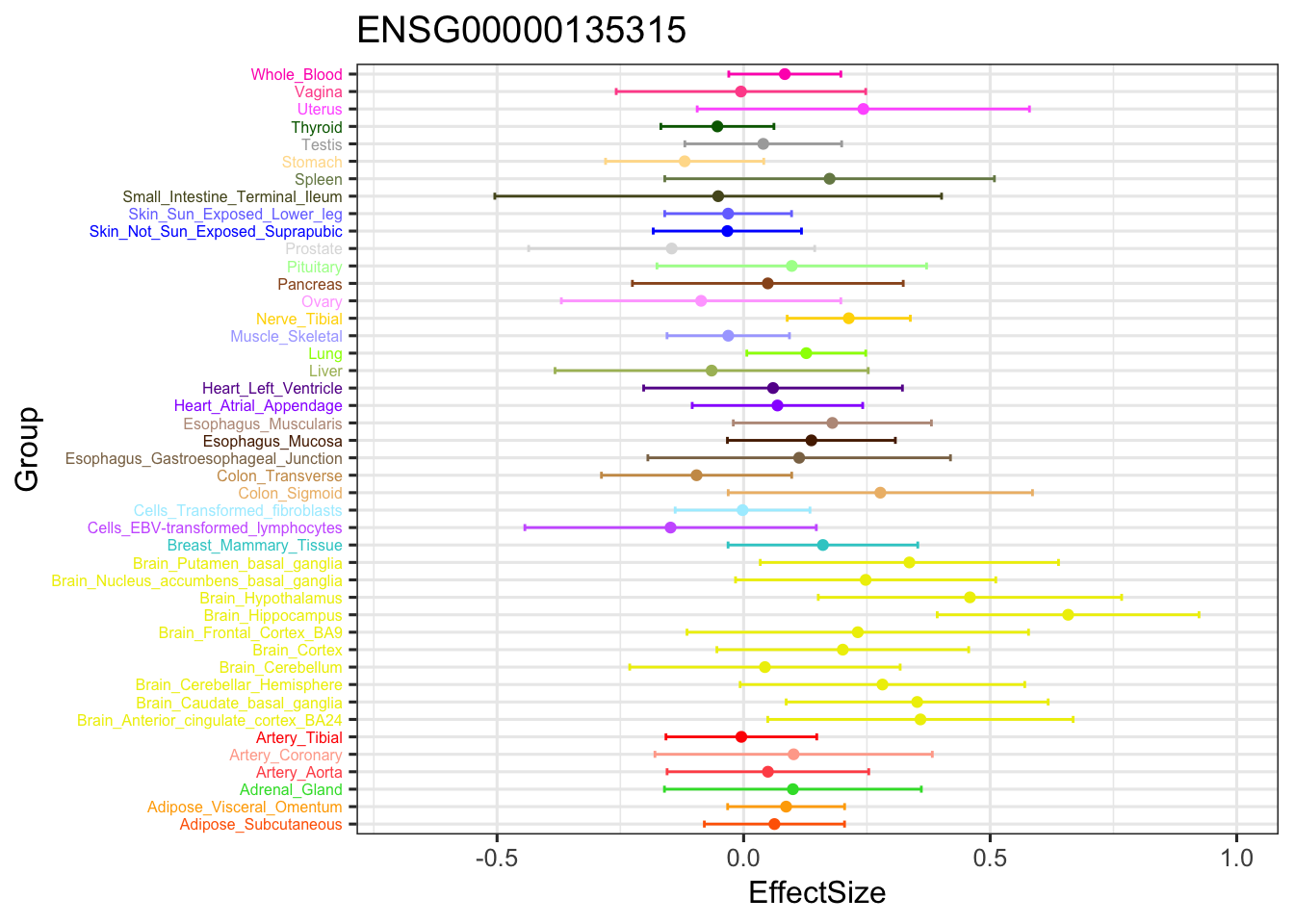
stronggeneSimple = data.frame(m_simple_EZ$result$PosteriorMean[5034,])
colnames(stronggeneSimple) = 'EffectSize'
stronggeneSimple$Group = row.names(stronggeneSimple)
stronggeneSimple$se = m_simple_EZ$result$PosteriorSD[5034,]
ggplot(stronggeneSimple, aes(y = EffectSize, x = Group)) +
geom_point(show.legend = FALSE, color=gtex.colors) + coord_flip() + ggtitle('ENSG00000135315 Simple') + ylim(c(-0.7,1)) +
geom_errorbar(aes(ymin=EffectSize-1.96*se, ymax=EffectSize+1.96*se), width=0.4, show.legend = FALSE, color=gtex.colors) +
theme_bw(base_size=12) + theme(axis.text.y = element_text(colour = gtex.colors, size = 6))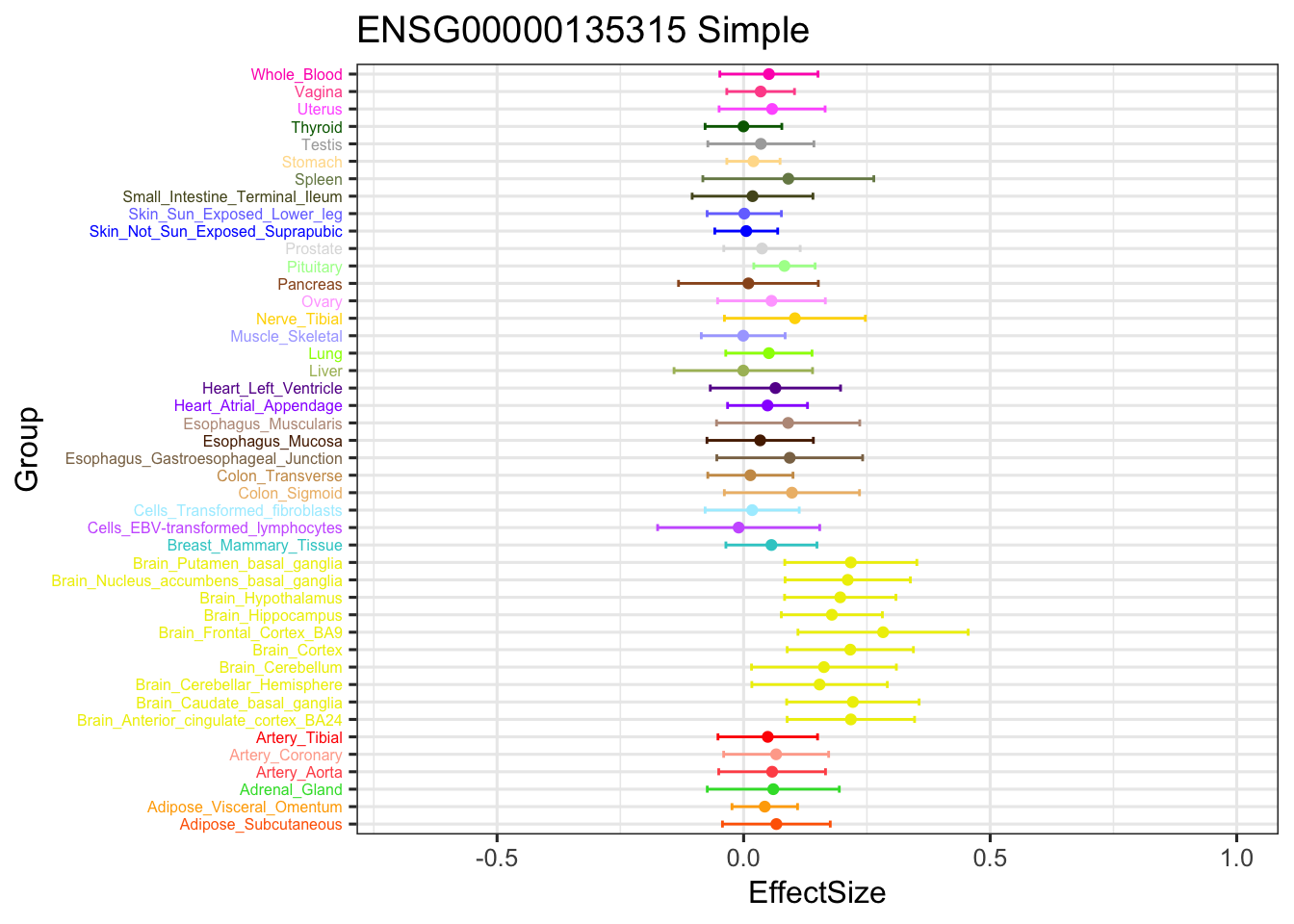
stronggeneCurrent = data.frame(m_current_EZ$result$PosteriorMean[5034,])
colnames(stronggeneCurrent) = 'EffectSize'
stronggeneCurrent$Group = row.names(stronggeneCurrent)
stronggeneCurrent$se = m_current_EZ$result$PosteriorSD[5034,]
ggplot(stronggeneCurrent, aes(y = EffectSize, x = Group)) +
geom_point(show.legend = FALSE, color=gtex.colors) + ylim(c(-0.7,1)) + coord_flip() + ggtitle('ENSG00000135315 Current') +
geom_errorbar(aes(ymin=EffectSize-1.96*se, ymax=EffectSize+1.96*se), width=0.4, show.legend = FALSE, color=gtex.colors) +
theme_bw(base_size=12) + theme(axis.text.y = element_text(colour = gtex.colors, size = 6))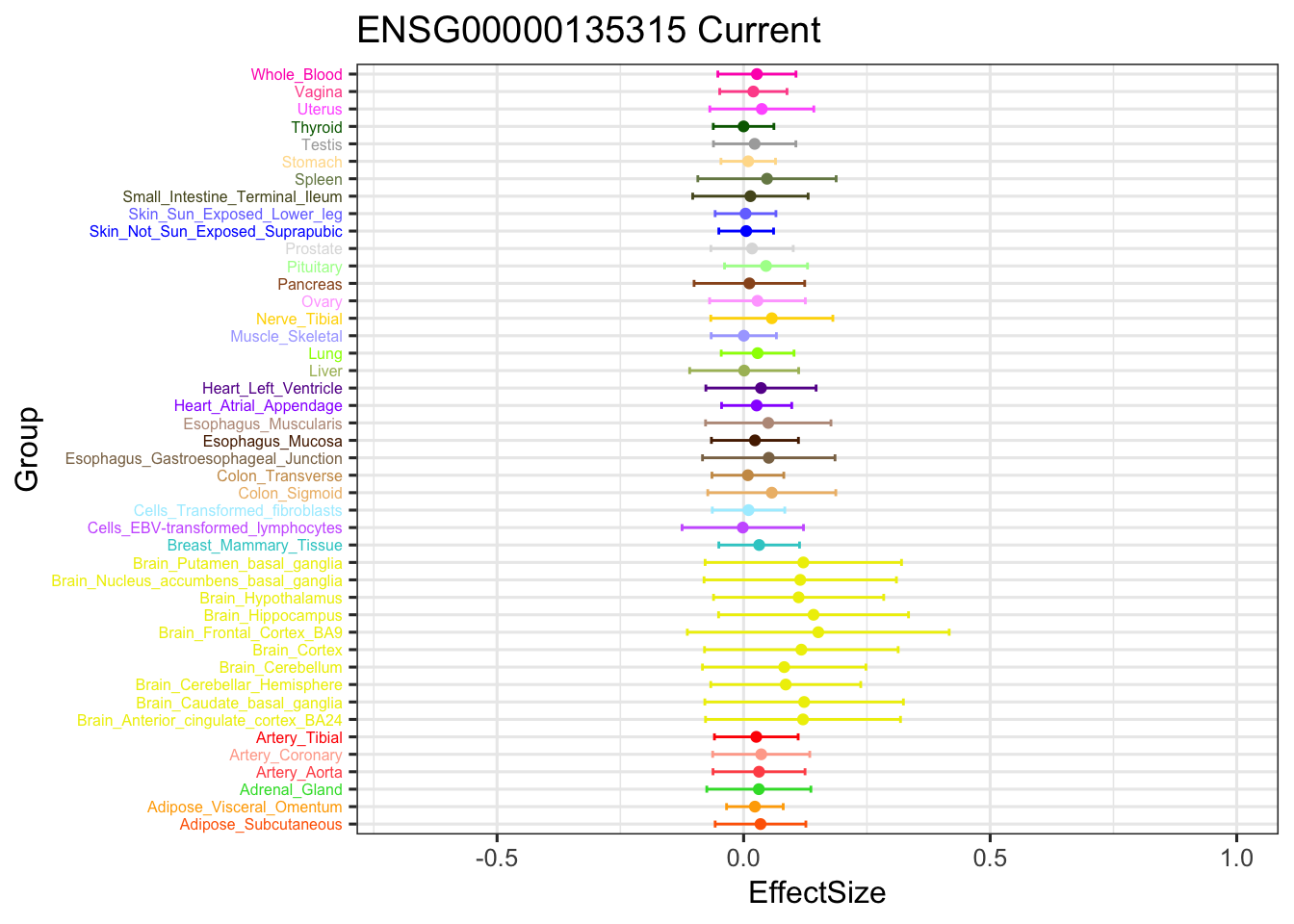
stronggeneV3 = data.frame(m_V3_EZ$result$PosteriorMean[5034,])
colnames(stronggeneV3) = 'EffectSize'
stronggeneV3$Group = row.names(stronggeneV3)
stronggeneV3$se = m_V3_EZ$result$PosteriorSD[5034,]
ggplot(stronggeneV3, aes(y = EffectSize, x = Group)) +
geom_point(show.legend = FALSE, color=gtex.colors) + ylim(c(-0.7,1)) + coord_flip() + ggtitle('ENSG00000135315 V3') +
geom_errorbar(aes(ymin=EffectSize-1.96*se, ymax=EffectSize+1.96*se), width=0.4, show.legend = FALSE, color=gtex.colors) +
theme_bw(base_size=12) + theme(axis.text.y = element_text(colour = gtex.colors, size = 6))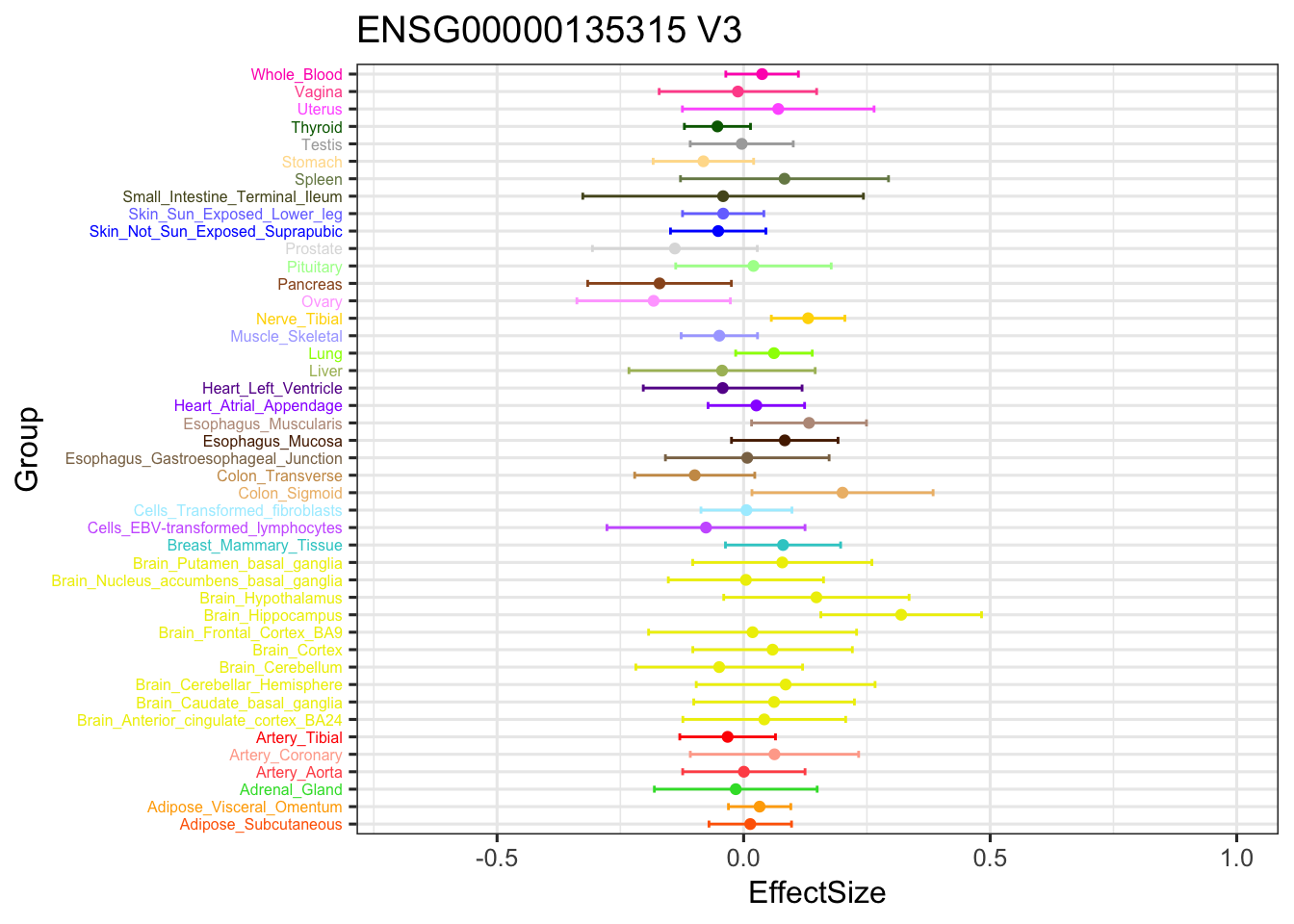
The gene MCPH1:
par(mfrow=c(1,3))
stronggene = data.frame(gtex$strong.b[13837,])
colnames(stronggene) = 'EffectSize'
stronggene$Group = row.names(stronggene)
stronggene$se = gtex$strong.s[13837,]
ggplot(stronggene, aes(y = EffectSize, x = Group)) +
geom_point(show.legend = FALSE, color=gtex.colors) + coord_flip() + ggtitle('ENSG00000249898') + ylim(c(-1.3,1.1)) + geom_errorbar(aes(ymin=EffectSize-1.96*se, ymax=EffectSize+1.96*se), width=0.4, show.legend = FALSE, color=gtex.colors) +
theme_bw(base_size=12) + theme(axis.text.y = element_text(colour = gtex.colors, size = 6))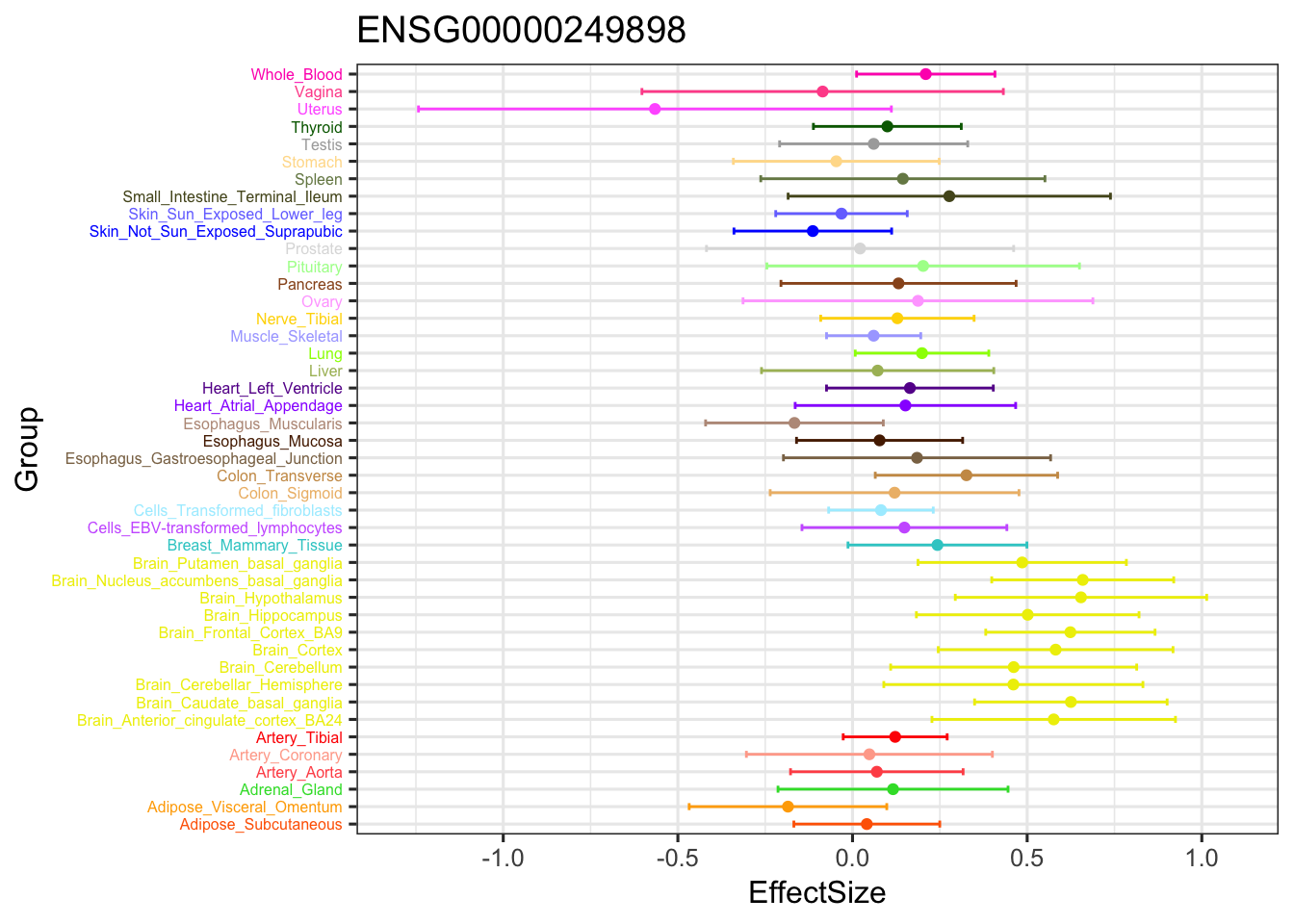
stronggeneCurrent = data.frame(m_current_EZ$result$PosteriorMean[13837,])
colnames(stronggeneCurrent) = 'EffectSize'
stronggeneCurrent$Group = row.names(stronggeneCurrent)
stronggeneCurrent$se = m_current_EZ$result$PosteriorSD[13837,]
ggplot(stronggeneCurrent, aes(y = EffectSize, x = Group)) +
geom_point(show.legend = FALSE, color=gtex.colors) + coord_flip() + ggtitle('ENSG00000249898 Current') + ylim(c(-1.3,1.1)) +
geom_errorbar(aes(ymin=EffectSize-1.96*se, ymax=EffectSize+1.96*se), width=0.4, show.legend = FALSE, color=gtex.colors) +
theme_bw(base_size=12) + theme(axis.text.y = element_text(colour = gtex.colors, size = 6))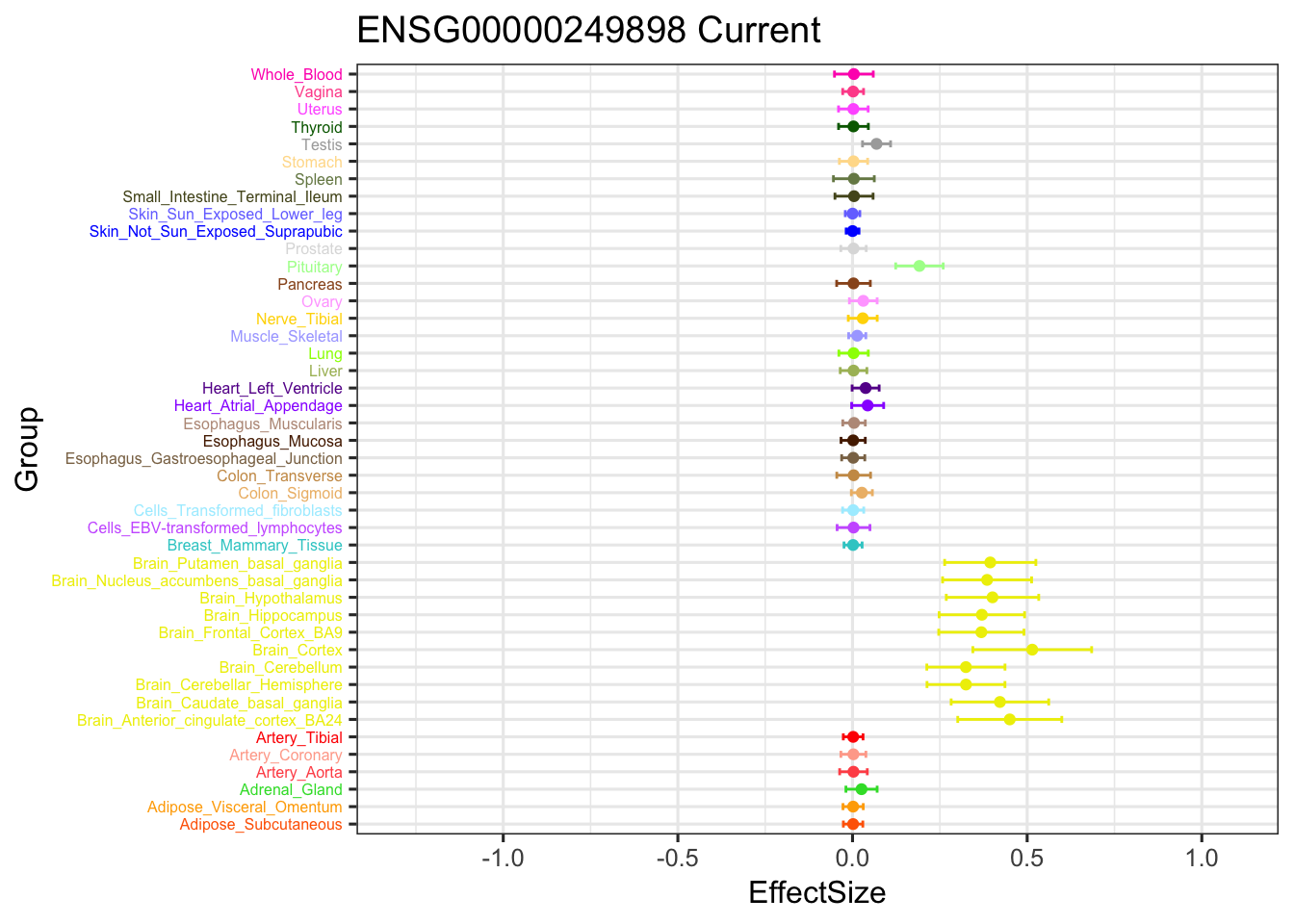
stronggeneV3 = data.frame(m_V3_EZ$result$PosteriorMean[13837,])
colnames(stronggeneV3) = 'EffectSize'
stronggeneV3$Group = row.names(stronggeneV3)
stronggeneV3$se = m_V3_EZ$result$PosteriorSD[13837,]
ggplot(stronggeneV3, aes(y = EffectSize, x = Group)) +
geom_point(show.legend = FALSE, color=gtex.colors) + ylim(c(-1.3,1.1)) + coord_flip() + ggtitle('ENSG00000249898 V3') +
geom_errorbar(aes(ymin=EffectSize-1.96*se, ymax=EffectSize+1.96*se), width=0.4, show.legend = FALSE, color=gtex.colors) +
theme_bw(base_size=12) + theme(axis.text.y = element_text(colour = gtex.colors, size = 6))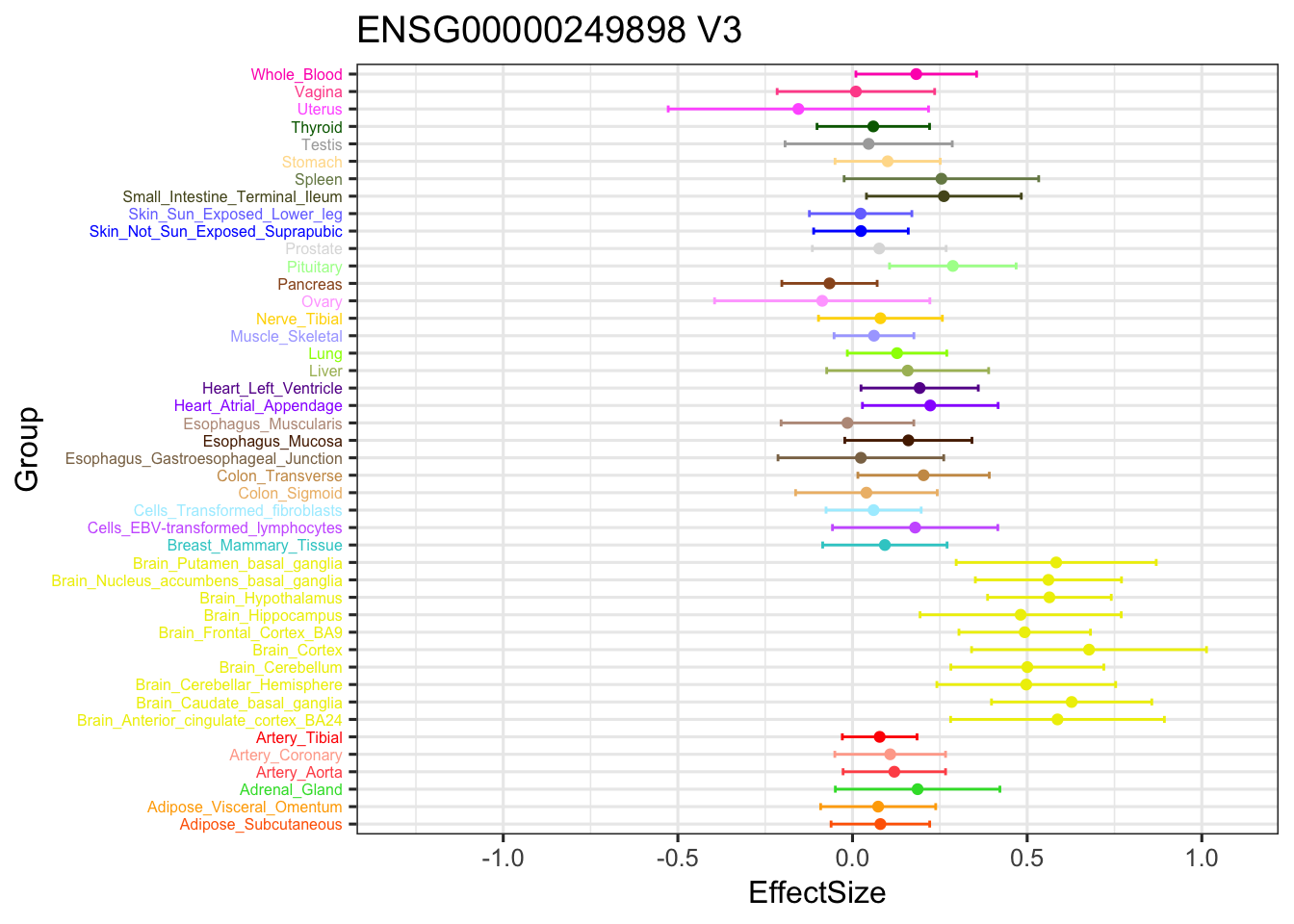
The gene significant in V3 EZ, not in current EZ:
ind = setdiff(get_significant_results(m_V3_EZ), get_significant_results(m_current_EZ))[1]
par(mfrow=c(1,3))
stronggene = data.frame(gtex$strong.b[ind,])
colnames(stronggene) = 'EffectSize'
stronggene$Group = row.names(stronggene)
stronggene$se = gtex$strong.s[ind,]
ggplot(stronggene, aes(y = EffectSize, x = Group)) +
geom_point(show.legend = FALSE, color=gtex.colors) + coord_flip() + ggtitle('ENSG00000126243') + ylim(c(-1.3,1.1)) + geom_errorbar(aes(ymin=EffectSize-1.96*se, ymax=EffectSize+1.96*se), width=0.4, show.legend = FALSE, color=gtex.colors) +
theme_bw(base_size=12) + theme(axis.text.y = element_text(colour = gtex.colors, size = 6))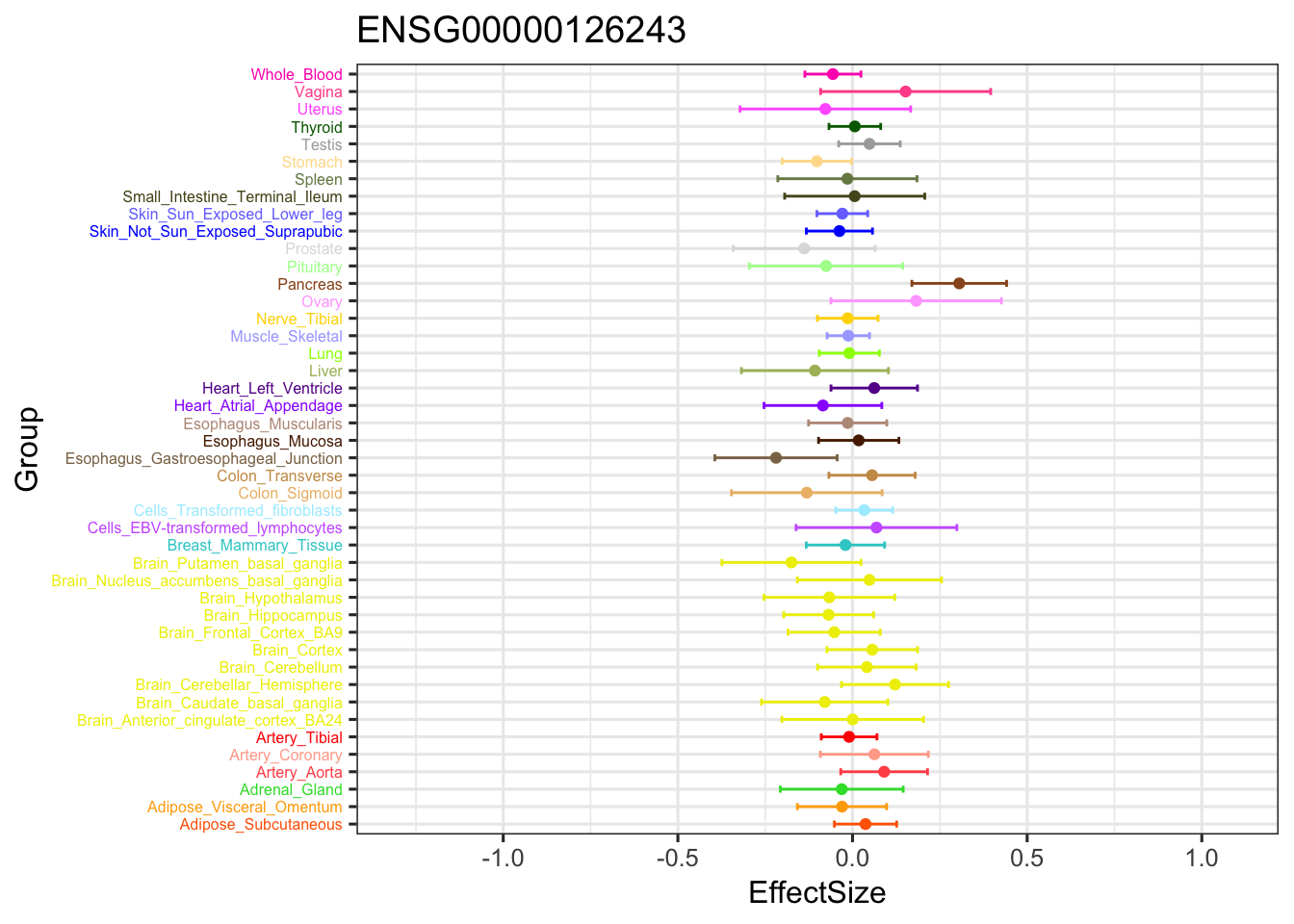
stronggeneCurrent = data.frame(m_current_EZ$result$PosteriorMean[ind,])
colnames(stronggeneCurrent) = 'EffectSize'
stronggeneCurrent$Group = row.names(stronggeneCurrent)
stronggeneCurrent$se = m_current_EZ$result$PosteriorSD[ind,]
ggplot(stronggeneCurrent, aes(y = EffectSize, x = Group)) +
geom_point(show.legend = FALSE, color=gtex.colors) + coord_flip() + ggtitle('ENSG00000126243 Current') + ylim(c(-1.3,1.1)) +
geom_errorbar(aes(ymin=EffectSize-1.96*se, ymax=EffectSize+1.96*se), width=0.4, show.legend = FALSE, color=gtex.colors) +
theme_bw(base_size=12) + theme(axis.text.y = element_text(colour = gtex.colors, size = 6))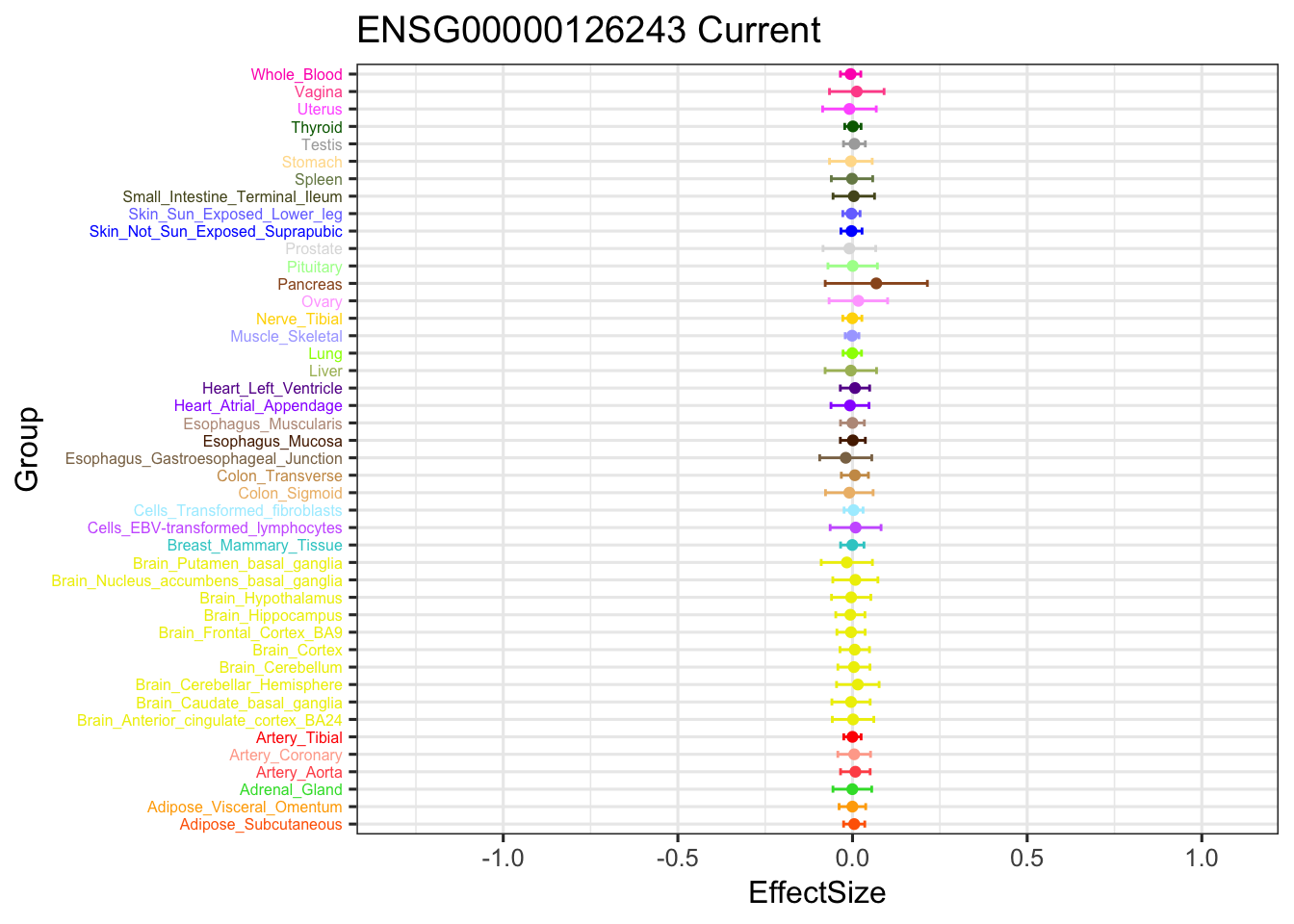
stronggeneV3 = data.frame(m_V3_EZ$result$PosteriorMean[ind,])
colnames(stronggeneV3) = 'EffectSize'
stronggeneV3$Group = row.names(stronggeneV3)
stronggeneV3$se = m_V3_EZ$result$PosteriorSD[ind,]
ggplot(stronggeneV3, aes(y = EffectSize, x = Group)) +
geom_point(show.legend = FALSE, color=gtex.colors) + ylim(c(-1.3,1.1)) + coord_flip() + ggtitle('ENSG00000126243 V3') +
geom_errorbar(aes(ymin=EffectSize-1.96*se, ymax=EffectSize+1.96*se), width=0.4, show.legend = FALSE, color=gtex.colors) +
theme_bw(base_size=12) + theme(axis.text.y = element_text(colour = gtex.colors, size = 6))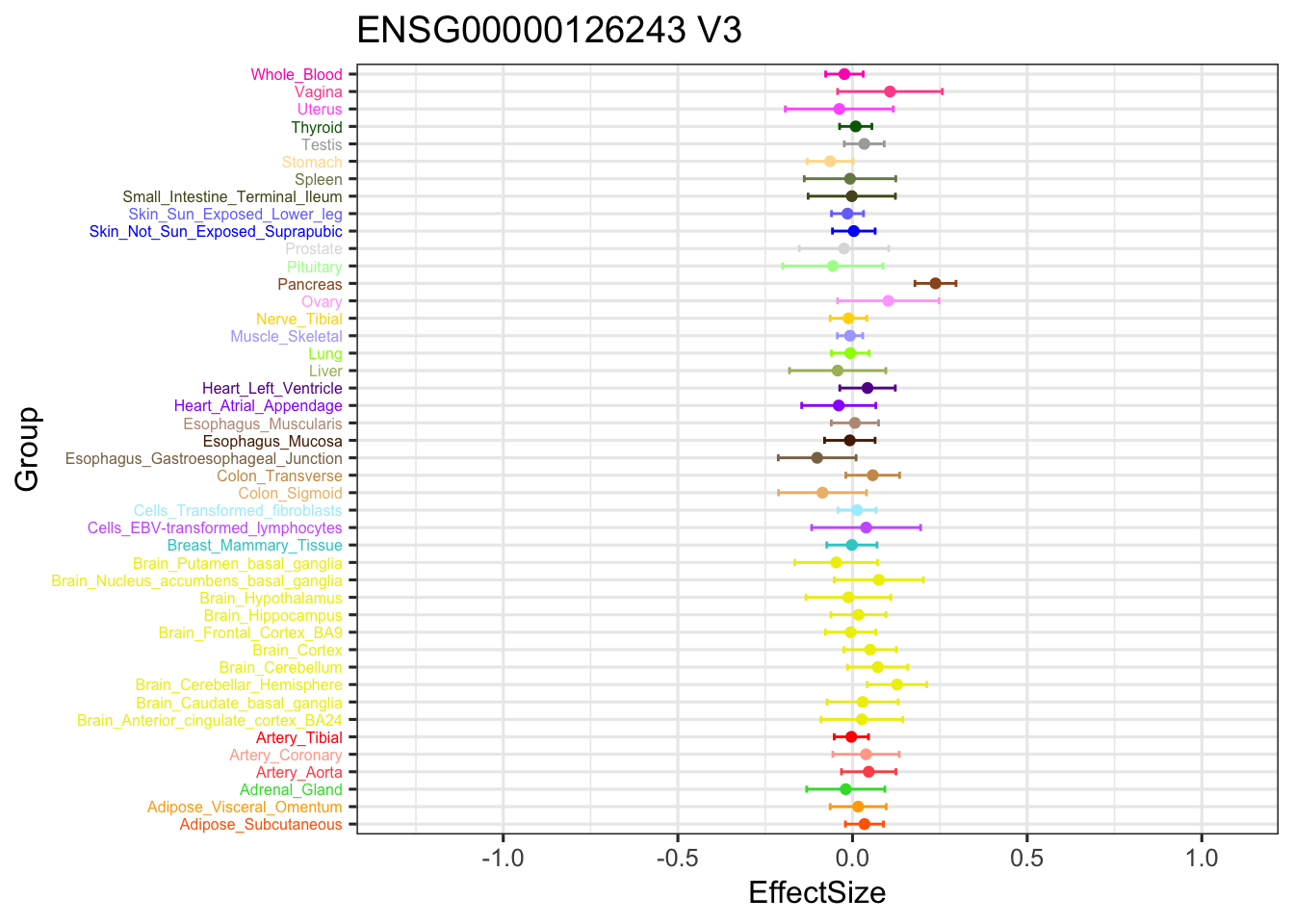
The pairwise sharing by magnitude
par(mfrow = c(1,2))
x <- get_pairwise_sharing(m_ignore_EZ)
colnames(x) <- colnames(get_lfsr(m_ignore_EZ))
rownames(x) <- colnames(x)
clrs=colorRampPalette(rev(c('darkred', 'red','orange','yellow','cadetblue1', 'cyan', 'dodgerblue4', 'blue','darkorchid1','lightgreen','green', 'forestgreen','darkolivegreen')))(200)
corrplot::corrplot(x, method='color', type='upper', tl.col="black", tl.srt=45, tl.cex = 0.7, diag = FALSE, col=clrs, cl.lim = c(0,1), title = 'Ignore EZ', mar=c(0,0,5,0))Warning in corrplot::corrplot(x, method = "color", type = "upper", tl.col =
"black", : Not been able to calculate text margin, please try again with a
clean new empty window using {plot.new(); dev.off()} or reduce tl.cexx <- get_pairwise_sharing(m_simple_EZ)
colnames(x) <- colnames(get_lfsr(m_simple_EZ))
rownames(x) <- colnames(x)
corrplot::corrplot(x, method='color', type='upper', tl.col="black", tl.srt=45, tl.cex = 0.7, diag = FALSE, col=clrs, cl.lim = c(0,1), title = 'Simple EZ', mar=c(0,0,5,0))Warning in corrplot::corrplot(x, method = "color", type = "upper", tl.col =
"black", : Not been able to calculate text margin, please try again with a
clean new empty window using {plot.new(); dev.off()} or reduce tl.cex
par(mfrow=c(1,2))
x <- get_pairwise_sharing(m_current_EZ)
colnames(x) <- colnames(get_lfsr(m_current_EZ))
rownames(x) <- colnames(x)
corrplot::corrplot(x, method='color', type='upper', tl.col="black", tl.srt=45, tl.cex = 0.7, diag = FALSE, col=clrs, cl.lim = c(0,1), title = 'Current EZ', mar=c(0,0,5,0))Warning in corrplot::corrplot(x, method = "color", type = "upper", tl.col =
"black", : Not been able to calculate text margin, please try again with a
clean new empty window using {plot.new(); dev.off()} or reduce tl.cexx <- get_pairwise_sharing(m_V3_EZ)
colnames(x) <- colnames(get_lfsr(m_V3_EZ))
rownames(x) <- colnames(x)
corrplot::corrplot(x, method='color', type='upper', tl.col="black", tl.srt=45, tl.cex = 0.7, diag = FALSE, col=clrs, cl.lim = c(0,1), title = 'V3 EZ', mar=c(0,0,5,0))Warning in corrplot::corrplot(x, method = "color", type = "upper", tl.col =
"black", : Not been able to calculate text margin, please try again with a
clean new empty window using {plot.new(); dev.off()} or reduce tl.cex
Session information
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.14.2
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggplot2_3.1.0 kableExtra_0.9.0 knitr_1.20 mclust_5.4.1
[5] mashr_0.2.19.0555 ashr_2.2-26 mixsqp_0.1-93 flashr_0.6-3
loaded via a namespace (and not attached):
[1] softImpute_1.4 tidyselect_0.2.5 corrplot_0.84
[4] purrr_0.2.5 reshape2_1.4.3 lattice_0.20-35
[7] colorspace_1.3-2 viridisLite_0.3.0 htmltools_0.3.6
[10] yaml_2.2.0 rlang_0.3.0.1 R.oo_1.22.0
[13] pillar_1.3.1 withr_2.1.2 glue_1.3.0
[16] R.utils_2.7.0 bindrcpp_0.2.2 foreach_1.4.4
[19] plyr_1.8.4 bindr_0.1.1 stringr_1.3.1
[22] munsell_0.5.0 gtable_0.2.0 workflowr_1.1.1
[25] rvest_0.3.2 R.methodsS3_1.7.1 mvtnorm_1.0-8
[28] codetools_0.2-15 evaluate_0.12 labeling_0.3
[31] pscl_1.5.2 doParallel_1.0.14 parallel_3.5.1
[34] highr_0.7 Rcpp_1.0.0 readr_1.1.1
[37] scales_1.0.0 backports_1.1.2 rmeta_3.0
[40] truncnorm_1.0-8 abind_1.4-5 hms_0.4.2
[43] digest_0.6.18 stringi_1.2.4 dplyr_0.7.6
[46] grid_3.5.1 rprojroot_1.3-2 tools_3.5.1
[49] magrittr_1.5 lazyeval_0.2.1 tibble_1.4.2
[52] crayon_1.3.4 whisker_0.3-2 pkgconfig_2.0.2
[55] MASS_7.3-50 Matrix_1.2-14 xml2_1.2.0
[58] SQUAREM_2017.10-1 httr_1.3.1 rstudioapi_0.8
[61] assertthat_0.2.0 rmarkdown_1.10 iterators_1.0.10
[64] R6_2.3.0 git2r_0.23.0 compiler_3.5.1 This reproducible R Markdown analysis was created with workflowr 1.1.1