2018Week7
Vicki Hertzberg
2/28/2018
Analyzing Microbiome Data
Many of you are here today because you have an interest in the microbiome, that “new organ” comprising the trillions of microbes that live in and on us. Today we are going to talk about how to identify these organisms isolated from a sample by sequencing a portion of the genetic material present, lining these up into counts of organisms, then using these data to summarize the microbes present in numeric or graphical fashion.
Before we get to that I just have one short message: (https://www.youtube.com/watch?v=htbeJhtFAXw)[https://www.youtube.com/watch?v=htbeJhtFAXw]
We will proceed as follows:
We will by briefly discussing the concept of metagenomics in general, and the 16S region of the bacterial genome in particular.
We will next discuss sequencing the .fastq file format. We are talking about .fastq files to give you an idea of what is coming out of the sequencer, why there is uncertainty, and why this is all so darned complicated.
We will walk through an example of processing a set of forward and reverse .fastq files to come up with something akin to an OTU table.
We will next use our “OTU table” together with data about our samples to produce numerical and visual descriptive summaries of our samples.
We will end by again using this “OTU table” and our sample data to develop functional profiles of our samples.
Metagenomics
Genomics, Proteomics, Transcriptomics
- These focus on the whole of a single individual organism.
- But organisms live in communities and interact.
- To study a community, we need to study across the genomes of the individual organisms comprising it.
Metagenomics
- The study of metagenomes, genetic material sampled directly from a community in its natural environment.
- Obviates the need for isolation and lab cultivation.
There are several methods for characterizing metagenomes. Some of these involve sequencing amplified markers, “amplicons” of eukaryotic genomes. For bacteria and archaeae, these are typically highly conserved small subunits of the gene that encodes for ribosomal RNA, in particular the 5S, 16S, 18Sm or 23S regions. For instance, the 16S rRNA unit, about 1500 base pairs in length, has 9 hypervariable regions. For fungi, the ITS unit serves this purpose. You can design an experiment so that you target one of those regions, or you can sequence the whole 16S region. Your choice of primers (and budget) will determine how much you sequence.
Alternatively you can perform whole genome sequencing (WGS), sometimes also called “shotgun” sequencing, to determine the whole bacterial genome. WGS is about 10 times more expensive then 16S sequencing.
16S rRNA
Universal PCR primers are used to amplify this region. Primers have been designed to match highly conserved regions of this subunit. A 2-D representation of the 16S region for the Thermus thermophilus, an extremophile bacteria, can be found at (http://rna.ucsc.edu/rnacenter/images/figs/thermus_16s_2ndry.jpg)[http://rna.ucsc.edu/rnacenter/images/figs/thermus_16s_2ndry.jpg]
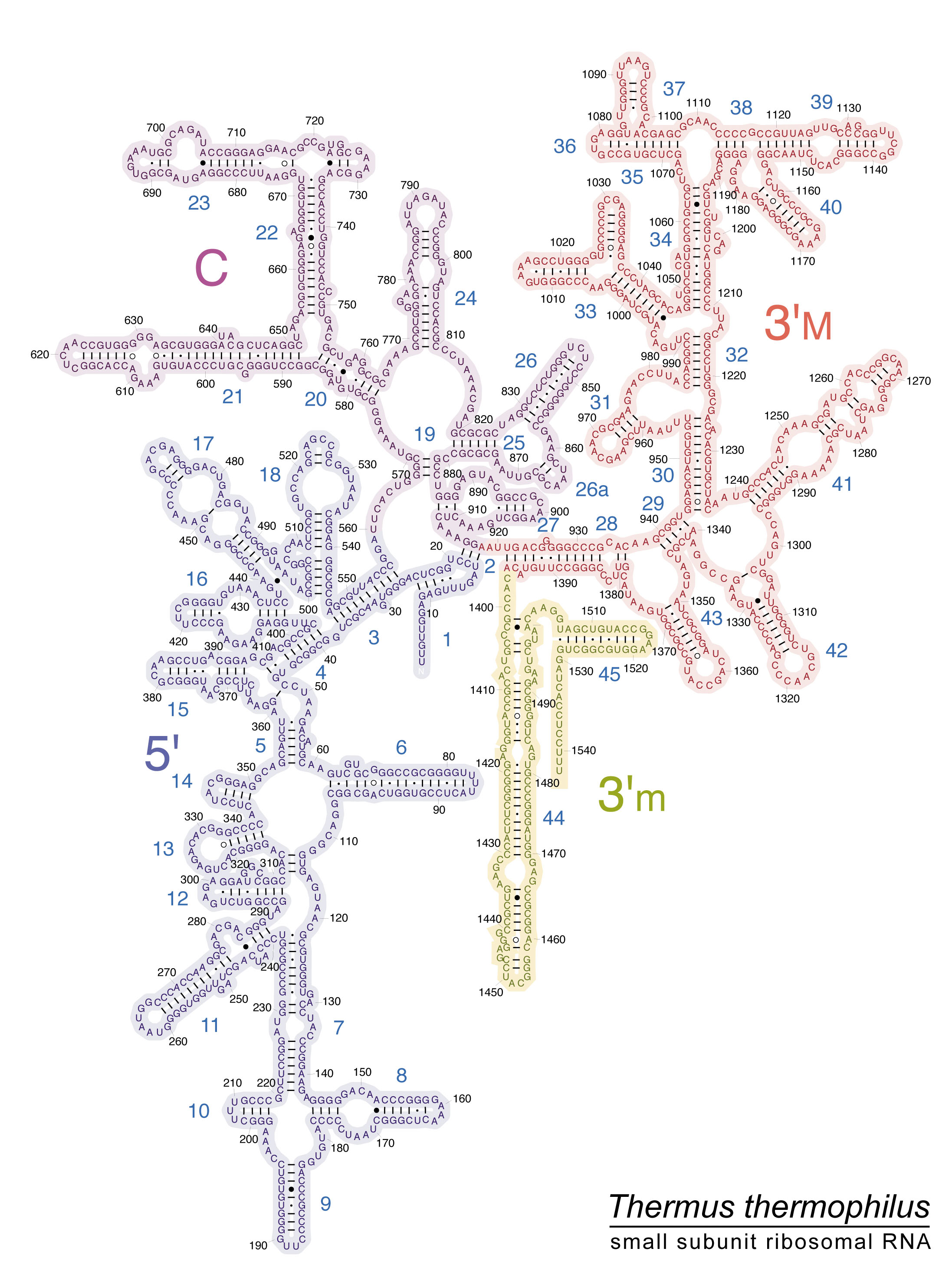{kind=link}
V4 Hypervariable Region
Universal primers:
- Forward (515F): GTGCCAGCMGCCGCGGTAA
- Reverse (805R): GGACTACHVGGGTWTCTAA
- These will produce 288 - 290 bp amplicons covering the V4 region
Sequences determined by Illumina MiSeq protocol
- Should get nearly complete overlap of forward and reverse reads.
- These can be merged to form a high quality consense base call at each position.
- Then classify the merged reads.
Wait! I thought that the only letters allowed in the sequences are A, C, G, or T. Yet I see M, H, V, and W in those primer sequences. What’s up with that?
It turns out that the Illumina is smart enough to know what it doesn’t know. So if it knows the base, it calls it that way. OTOH, if it can only resolve the base call down to, say, the choice of 2 bases, it uses these other letters to designate that situation. These are called the IUPAC Ambiguity Codes, and you can see them here: (http://droog.gs.washington.edu/parc/images/iupac.html)[http://droog.gs.washington.edu/parc/images/iupac.html]. There is another IUPAC code not included on the table, and that is “-”, which is used to indicate a gap, an often useful indicator for alignments.
Sequencing File Format
In the files produced by the sequencer, every read represents an independent copy of source DNA in your sample. When the target material is sequenced, there are two main considerations: sequencing breadth and sequencing depth.
- Breadth refers to the extent to which you sequence the entire genome present. You want to be sure that you have sequence information for all areas of the target.
Depth refers to how many reads on average cover each base pair of your target. This is also sometimes referred to as “coverage.”
- In 16s rRNA amplicon sequencing, the primers that you use determine breadth.
If you don’t have sufficient depth you may end up with incomplete or inconclusive results. OTOH, oversequencing raises costs.
Illumina FASTQ Files
File Naming Conventions:
- NA10831_ATCACG_L002_R1_001.fastq.gz
- FA1_S1_L001_R1_001.fastq.gz
- Sample_Barcode/Index_Lane_Read#_Set#.fastq.gz
Sequence Identifiers
- @EAS139:136:FC706VJ:2:5:1000:12850 1:N:18:ATCACG
- @M00763:36:000000000-A8T0A:1:1101:14740:1627 1:N:0:1
- @Instrument : Run# : FlowcellID : Lane : Tile : X : Y Read : Filtered : Control# : Barcode/Index
FASTQ File Format
| 4 lines per read as follows: |
|---|
| @sequence_id |
| sequence |
| + |
| quality |
| —————————— |
Example
Line1: @M00763:36:000000000-A8T0A:1:1101:14740:1627 1:N:0:1
Line2: CCTACGGGAGGCAGCAGTGGGGAATATTGCACAATGGGGGAAACCCTGATGC AGCGACGCCGCGTGAGTGAAGAAGTATCTCGGTATGTAAAGCTCTATCAGCA GGAAAGATAATGACGGTACCTGACTAAGAAGCCCCGGCTAACTACGTGCCAG CAGCCGCGGTAATACGTAGGGGGCAAGCGTTATCCGGATTTACTGGGTGTAA AGGGAGCGTAGACGGCAGCGCAAGTCTGGAGTGAAATGCCGGGGCCCAACCC CGGCCCTGCTTTGGAACCCGTCCCGCTCCAGTGCGGGCGGG
Line3: +
Line 4: 88CCCGDBAF)===CEFFGGGG>GGGGGGCCFGGGGGDFGGGGDCFGGGFED CFG:@CFCGGGGGGG?FFG9FFFGG9ECEFGGGDFGGGFFEFAFAFFEFECE F@4AFD85CFFAA?7+C@FFF<,A?,,,,,,AFFF77BFC,8>,>8D@FFFF G,ACGGGCFG>*57;*6=C58:?<)9?:=:C*;;@C?3977@C7E*;29>/= +2**)75):17)8@EE3>D59>)>).)61)4>(6*+/)@F63639993??D1 :0)((,((.(.+)(()(-(*-(-((-,,(.(.)),(-0)))
Quality Scores
The 4th line consists of the Phred scores for each base call. Each character is associated with a value between 0 and 40, and these are called the Quality scores or Q scores. The coding scheme can be found at (https://support.illumina.com/help/BaseSpace_OLH_009008/Content/Source/Informatics/BS/QualityScoreEncoding_swBS.htm)[https://support.illumina.com/help/BaseSpace_OLH_009008/Content/Source/Informatics/BS/QualityScoreEncoding_swBS.htm]
The relationship is as follows:
\[\begin{equation} Q=-10*log(p) \end{equation}\]Or you can solve for p as follows:
\[\begin{equation} p=10^{-Q/10} \end{equation}\]You can see from the table that letters have high Quality Scores. Let’s look at the Phred score of I, which has a Q value of 40.
| Q=40 |
| -Q/10 = -4 |
| p = \(10^{-4}\) |
| p = 0.0001 |
In contrast, consider when the Phred score is “+”, the Q value is 10, and p = 0.10.
In summary, if you are looking at the .fastq file (which you can open with a text editor), letters are good, numbers are medium, and most anything else is of lesser quality.
You want to know this because you may want to trim your sequences. Sometimes when a sequence is read, the first 10 or so sequences are of lesser quality. Also, reads deteriorate in quality towards the end of a sequence. Finally, forward reads are generally of better quality than their corresponding reverse reads.
Processing Sequencing Data: Overview
- Filter - remove low-quality reads and non-target sequences
- Trim - prune low-quality ends
- Assembly - correct overlapping bases
- Aggregate - combine similar reads
- Chimeras - remove chimeric reads
Processing Sequencing Data with the DADA2 Pipeline
Today we are going to go through the process of “feeding” a set of Illumina-sequenced paired-end .fastq files into the dada2 package, with the goal of deriving a sequence table in the end. The term “dada” stands for Divisive Amplicon Denoising Algorithm. This R package example uses 4 dada steps, 2 for the forward reads and 2 for the reverse reads, hence “dada2.”
A sequence table is akin to the ubiquitous OTU table, only it is at a much higher resolution. OTUs, or operational taxonomic units, is an operational term defined as sets of individual organisms that have been put together in groups according to their DNA similarity. OTUs are often used as proxies for “species”. “Similarity” in this context is defined as differing by no more than a fixed dissimilarity threshold, typically set at 3%. But we are going to do better than 3% dissimilarity; we are headed to 100%.
Why isn’t 97% good enough?
- It still is low resolution, fiving genus level identification at best
- It tends to have a high false positive rate, with the number of OTUs much greater than actual richness.
- As you get more samples, the processing time scales up super-linearly.
We are using dada2 because it infers sample sequences exactly, resolving differences of as little as one (1) nucleotide. Not only does dada2 provide higher resolution, it also performs better than two other major players in this field: mothur and QIIME (v1). But note that QIIME v2.1 now has a DADA2 step built into it, so it should now compete well with this package.
For the moment, however, we are going to continue to use dada2, well, because. Because we are now expert R, RStudio, git and github users, that’s why!!!
First Note of Importance
Our “input” into the package is a set of Illumina-sequenced paired-end .fastq files that have been demultiplexed (i.e., split by sample) and from which the barcodes/adapters/primers have already been removed. In this toy dataset all of these barcoode/adapters/primers have been removed. But when it comes to processing your own data, you will have to check this before you proceed.
My personal experience to with two different labs is that you will receive demultiplexed files with the barcodes removed, but the primers have not been removed. You will need to know that the primers are so that you can figure out the lengths. You can trim before hand with a program such as TRIMMOMATIC (http://www.usadellab.org/cms/?page=trimmomatic)[http://www.usadellab.org/cms/?page=trimmomatic]. This way, when we display the quality plots later you will be seeing the quality of the “meat” of your data, instead of having some distraction with the quality of the sequences including the primers.
You can also trim later by setting an option. I will show you (outside of an R chunk of course, because these data have already had the primers trimmed) how to do that.
Second Note of Importance
This pipeline also assumes that if you have paired reads, that the forward and reverse .fastq files contain the reads in matching order.
If this is not true, you will need to remedy this before proceeding. Please see the dada2 FAQ for suggestions: (http://benjjneb.github.io/dada2/faq.html#what-if-my-forward-and-reverse-reads-arent-in-matching-order)[http://benjjneb.github.io/dada2/faq.html#what-if-my-forward-and-reverse-reads-arent-in-matching-order].
End of Notes of Importance. For the Moment.
So let’s get started now.
Are We Ready?
Let’s check that all is ready.
library(dada2); packageVersion("dada2")## Loading required package: Rcpp## [1] '1.6.0'library(ShortRead); packageVersion("ShortRead")## Loading required package: BiocGenerics## Loading required package: parallel##
## Attaching package: 'BiocGenerics'## The following objects are masked from 'package:parallel':
##
## clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
## clusterExport, clusterMap, parApply, parCapply, parLapply,
## parLapplyLB, parRapply, parSapply, parSapplyLB## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs## The following objects are masked from 'package:base':
##
## anyDuplicated, append, as.data.frame, cbind, colMeans,
## colnames, colSums, do.call, duplicated, eval, evalq, Filter,
## Find, get, grep, grepl, intersect, is.unsorted, lapply,
## lengths, Map, mapply, match, mget, order, paste, pmax,
## pmax.int, pmin, pmin.int, Position, rank, rbind, Reduce,
## rowMeans, rownames, rowSums, sapply, setdiff, sort, table,
## tapply, union, unique, unsplit, which, which.max, which.min## Loading required package: BiocParallel## Loading required package: Biostrings## Loading required package: S4Vectors## Loading required package: stats4##
## Attaching package: 'S4Vectors'## The following object is masked from 'package:base':
##
## expand.grid## Loading required package: IRanges## Loading required package: XVector##
## Attaching package: 'Biostrings'## The following object is masked from 'package:base':
##
## strsplit## Loading required package: Rsamtools## Loading required package: GenomeInfoDb## Loading required package: GenomicRanges## Loading required package: GenomicAlignments## Loading required package: SummarizedExperiment## Loading required package: Biobase## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.## Loading required package: DelayedArray## Loading required package: matrixStats##
## Attaching package: 'matrixStats'## The following objects are masked from 'package:Biobase':
##
## anyMissing, rowMedians##
## Attaching package: 'DelayedArray'## The following objects are masked from 'package:matrixStats':
##
## colMaxs, colMins, colRanges, rowMaxs, rowMins, rowRanges## The following object is masked from 'package:Biostrings':
##
## type## The following object is masked from 'package:base':
##
## apply## [1] '1.36.1'library(phyloseq); packageVersion("phyloseq")##
## Attaching package: 'phyloseq'## The following object is masked from 'package:SummarizedExperiment':
##
## distance## The following object is masked from 'package:Biobase':
##
## sampleNames## The following object is masked from 'package:GenomicRanges':
##
## distance## The following object is masked from 'package:IRanges':
##
## distance## [1] '1.22.3'library(ggplot2); packageVersion("ggplot2")## [1] '2.2.1'I have also downloaded the file used in the Mothur MiSeq SOP, as well as two RDP reference files. The Mothur MiSeq files contain data from an experiment in which the V4 region of the 16S rRNA gene in mice feces was sequenced. You will have to change the path in the next chunk to the path to where your files sit. Also if you are on a Windows machine, this will also look different. Let’s make sure they are all in the proper place on my machine:
# Set the path to the data files
path <- "~/Documents/NRSG_741/MiSeqData/MiSeq_SOP"
fileNames <- list.files(path)
fileNames## [1] "F3D0_S188_L001_R1_001.fastq"
## [2] "F3D0_S188_L001_R2_001.fastq"
## [3] "F3D1_S189_L001_R1_001.fastq"
## [4] "F3D1_S189_L001_R2_001.fastq"
## [5] "F3D141_S207_L001_R1_001.fastq"
## [6] "F3D141_S207_L001_R2_001.fastq"
## [7] "F3D142_S208_L001_R1_001.fastq"
## [8] "F3D142_S208_L001_R2_001.fastq"
## [9] "F3D143_S209_L001_R1_001.fastq"
## [10] "F3D143_S209_L001_R2_001.fastq"
## [11] "F3D144_S210_L001_R1_001.fastq"
## [12] "F3D144_S210_L001_R2_001.fastq"
## [13] "F3D145_S211_L001_R1_001.fastq"
## [14] "F3D145_S211_L001_R2_001.fastq"
## [15] "F3D146_S212_L001_R1_001.fastq"
## [16] "F3D146_S212_L001_R2_001.fastq"
## [17] "F3D147_S213_L001_R1_001.fastq"
## [18] "F3D147_S213_L001_R2_001.fastq"
## [19] "F3D148_S214_L001_R1_001.fastq"
## [20] "F3D148_S214_L001_R2_001.fastq"
## [21] "F3D149_S215_L001_R1_001.fastq"
## [22] "F3D149_S215_L001_R2_001.fastq"
## [23] "F3D150_S216_L001_R1_001.fastq"
## [24] "F3D150_S216_L001_R2_001.fastq"
## [25] "F3D2_S190_L001_R1_001.fastq"
## [26] "F3D2_S190_L001_R2_001.fastq"
## [27] "F3D3_S191_L001_R1_001.fastq"
## [28] "F3D3_S191_L001_R2_001.fastq"
## [29] "F3D5_S193_L001_R1_001.fastq"
## [30] "F3D5_S193_L001_R2_001.fastq"
## [31] "F3D6_S194_L001_R1_001.fastq"
## [32] "F3D6_S194_L001_R2_001.fastq"
## [33] "F3D7_S195_L001_R1_001.fastq"
## [34] "F3D7_S195_L001_R2_001.fastq"
## [35] "F3D8_S196_L001_R1_001.fastq"
## [36] "F3D8_S196_L001_R2_001.fastq"
## [37] "F3D9_S197_L001_R1_001.fastq"
## [38] "F3D9_S197_L001_R2_001.fastq"
## [39] "filtered"
## [40] "HMP_MOCK.v35.fasta"
## [41] "Mock_S280_L001_R1_001.fastq"
## [42] "Mock_S280_L001_R2_001.fastq"
## [43] "mouse.dpw.metadata"
## [44] "mouse.time.design"
## [45] "raw quality plots"
## [46] "rdp_species_assignment_14.fa.gz"
## [47] "rdp_train_set_14.fa"
## [48] "rdp_train_set_14.fa.gz"
## [49] "silva_nr_v132_train_set.fa.gz"
## [50] "silva_species_assignment_v132.fa.gz"
## [51] "stability.batch"
## [52] "stability.files"OK, I see 38 .fastq files and the two SILVA V132 files. With the exception of the two SILVA files (which we list but the dada2 tutorial does not), we agree. The file named “filtered” will be created in another couple of steps, so we are not going to worry about that.
Filter and Trim
So now we are ready to use the dada2 pipeline. We will first read in the names of the .fastq files. Then we will manipulate those names as character variables, using regular expressions to create lists of the forward and reverse read .fastq files in matched order.
# Forward and reverse fastq filenames have format: SAMPLENAME_R1_001.fastq and SAMPLENAME_R2_001.fastq
# Read in the names of the .fastq files
fnFs <- sort(list.files(path, pattern="_R1_001.fastq", full.names=TRUE))
fnRs <- sort(list.files(path, pattern="_R2_001.fastq", full.names=TRUE))
# Extract sample names, assuming filenames have format: SAMPLENAME_XXX.fastq
sample.names <- sapply(strsplit(basename(fnFs), "_"), `[`, 1)Important Note 3
If you are using this workflow with your own data, you will probably need to modify the R chunk above, especially the assignment of sample names to the variable sample.names.
End of Note
Quality Profiles of the Reads
One of the points that we have repeatedly emphasized in this class is the importance of visualizing your data, and that process is still important with this type of data. Fortunately there is a great quality profile plot that you can generate with just a single command from dada2.
# Visualize the quality profile of the first two files containing forward reads
plotQualityProfile(fnFs[1:2])
We see here that the forward reads are really good quality. Callahan advises “trimming the last few nucleotides to avoid less well-controlled errors that can arise there.” OTOH, Christopher Taylor, who runs the Metagenomics lab at LSU Health Sciences Center advises to always trim the first 10 reads.
Let’s look at the reverse reads.
# Visualize the quality profile of the first two files containing reverse reads
plotQualityProfile(fnRs[1:2])
The quality of the reverse reads is subtantially worse, especially toward the end, a common phenomenon with Illumina paired-end sequencing. The dada algorithm incorporates error quality into the model, so it is robust to lower quality sequences, but trimming is still a good idea.
If you are using your own data, make sure that you have good overlap, the more the better.
Performing the Filtering and Trimming
We will use typical filtering parameters.
maxN = 0–dada2requires that there be no N’s in a sequencetruncQ = 2– truncate reads at the first instance of a quality less than or equal to #.maxEE= 2 – sets the maximum number of expected errors allowed in a read, which is a better filter than simply averaging quality scores.
Let’s jointly filter the forward and reverse reads with the fastqPairedFilter function.
# Make a directory and filenames for the filtered fastqs
# Place filtered files in a filtered/ subdirectory
filt.path <- file.path(path, "filtered")
if(!file_test("-d", filt.path)) dir.create(filt.path)
filtFs <- file.path(filt.path, paste0(sample.names, "_F_filt.fastq.gz"))
filtRs <- file.path(filt.path, paste0(sample.names, "_R_file.fastq.gz"))Now filter the forward and reverse reads
out <- filterAndTrim(fnFs, filtFs, fnRs, filtRs, truncLen = c(240, 160),
maxN=0, maxEE =c(2,2), truncQ = 2, rm.phix = TRUE,
compress=TRUE, multithread=TRUE) #On Windows set multithread=FALSE
head(out)## reads.in reads.out
## F3D0_S188_L001_R1_001.fastq 7793 7113
## F3D1_S189_L001_R1_001.fastq 5869 5299
## F3D141_S207_L001_R1_001.fastq 5958 5463
## F3D142_S208_L001_R1_001.fastq 3183 2914
## F3D143_S209_L001_R1_001.fastq 3178 2941
## F3D144_S210_L001_R1_001.fastq 4827 4312Important Note 4
Standard filtering parameters as shown here are guidelines, i.e., they are not set in stone. For example, if too few reads are passing the filter, consider relaxing maxEE, perhaps especially on the reverse reads, (e.g., maxEE=c(2,5)). If you want to speed up downstream computation and have fewer reads pass the filter, consider tightening maxEE (e.g., maxEE=c(1,1)). For paired-end reads consider the length of your amplicon when choosing truncLen as you reads MUST OVERLAP after truncation in order to merge later.
End of Note
Learn the Error Rates
The dada2 algorithm uses a parametric error model (err), and, of course, the amplicon dataset will have different error rates. The algorithm will learn its error model from the data by alternating estimation of error rates and composition of the sample until convergence of the sample on a jointly consistent solution (like the EM algorithm, if you happen to know that) (and if you don’t, it does not matter).
So we will run this joint inference 4 times. The first passes will be through the forward and reverse reads setting selfConsist = TRUE. The second passes will be through the forward and reverse reads with the learned error structure. On the first pass, the algorithm starts with an initial guess, which is that the maximum possible error rates in these data, that is, the error rates if only the most abundant sequence is correct, and all the rest are errors. This is what happens when we set err=NULL.
Let’s take a 5 minute break while we take the first pass through the Forward reads then the Reverse reads:
errF <- learnErrors(filtFs, multithread = TRUE)## Initializing error rates to maximum possible estimate.
## Sample 1 - 7113 reads in 1979 unique sequences.
## Sample 2 - 5299 reads in 1639 unique sequences.
## Sample 3 - 5463 reads in 1477 unique sequences.
## Sample 4 - 2914 reads in 904 unique sequences.
## Sample 5 - 2941 reads in 939 unique sequences.
## Sample 6 - 4312 reads in 1267 unique sequences.
## Sample 7 - 6741 reads in 1756 unique sequences.
## Sample 8 - 4560 reads in 1438 unique sequences.
## Sample 9 - 15637 reads in 3590 unique sequences.
## Sample 10 - 11413 reads in 2762 unique sequences.
## Sample 11 - 12017 reads in 3021 unique sequences.
## Sample 12 - 5032 reads in 1566 unique sequences.
## Sample 13 - 18075 reads in 3707 unique sequences.
## Sample 14 - 6250 reads in 1479 unique sequences.
## Sample 15 - 4052 reads in 1195 unique sequences.
## Sample 16 - 7369 reads in 1832 unique sequences.
## Sample 17 - 4765 reads in 1183 unique sequences.
## Sample 18 - 4871 reads in 1382 unique sequences.
## Sample 19 - 6504 reads in 1709 unique sequences.
## Sample 20 - 4314 reads in 897 unique sequences.
## selfConsist step 2
## selfConsist step 3
## selfConsist step 4
## selfConsist step 5
## Convergence after 5 rounds.
## Total reads used: 139642errR <- learnErrors(filtRs, multithread = TRUE)## Initializing error rates to maximum possible estimate.
## Sample 1 - 7113 reads in 1660 unique sequences.
## Sample 2 - 5299 reads in 1349 unique sequences.
## Sample 3 - 5463 reads in 1335 unique sequences.
## Sample 4 - 2914 reads in 853 unique sequences.
## Sample 5 - 2941 reads in 880 unique sequences.
## Sample 6 - 4312 reads in 1286 unique sequences.
## Sample 7 - 6741 reads in 1803 unique sequences.
## Sample 8 - 4560 reads in 1265 unique sequences.
## Sample 9 - 15637 reads in 3414 unique sequences.
## Sample 10 - 11413 reads in 2522 unique sequences.
## Sample 11 - 12017 reads in 2771 unique sequences.
## Sample 12 - 5032 reads in 1415 unique sequences.
## Sample 13 - 18075 reads in 3290 unique sequences.
## Sample 14 - 6250 reads in 1390 unique sequences.
## Sample 15 - 4052 reads in 1134 unique sequences.
## Sample 16 - 7369 reads in 1635 unique sequences.
## Sample 17 - 4765 reads in 1084 unique sequences.
## Sample 18 - 4871 reads in 1161 unique sequences.
## Sample 19 - 6504 reads in 1502 unique sequences.
## Sample 20 - 4314 reads in 732 unique sequences.
## selfConsist step 2
## selfConsist step 3
## selfConsist step 4
## selfConsist step 5
## selfConsist step 6
## Convergence after 6 rounds.
## Total reads used: 139642Finally it is always worthwhile to visualize the estimated error rates:
# Plot the estimated error rates for the Forward reads
plotErrors(errF, nominalQ=TRUE)## Warning: Transformation introduced infinite values in continuous y-axis
## Warning: Transformation introduced infinite values in continuous y-axis
# And for the Reverse reads
plotErrors(errR, nominalQ = TRUE)## Warning: Transformation introduced infinite values in continuous y-axis
## Warning: Transformation introduced infinite values in continuous y-axis
The error for each possible type of transition (i.e., A -> C, A -> T, …, T -> G) are shown. The black points are the observed error rates for each consensus quality score. The black line shows the estimated error rates after convergence. The red line is the error rates expected under the nominal definition of the Q value. You see that the black line (estimated rates) fots the observed rates well, and the error rates drop with increased quality as expected. So all is looking good and we proceed.
Dereplication
You can gain further efficiencies by dereplicating the reads, ths is combining all identical sequences so that all you are left with is a list of “unique sequences” and a count of them, defined as the “abundance”. Other pipelines can do this too to gain efficiency, but dada2 retains a summary of the quality information associated with each unique sequence, developing a consensus quality profile as the average of the positional qualities from the dereplicated reads, which it then uses to inform the error model in the subsequent denoising step.
# Dereplicate
derepFs <- derepFastq(filtFs, verbose=TRUE)## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D0_F_filt.fastq.gz## Encountered 1979 unique sequences from 7113 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D1_F_filt.fastq.gz## Encountered 1639 unique sequences from 5299 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D141_F_filt.fastq.gz## Encountered 1477 unique sequences from 5463 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D142_F_filt.fastq.gz## Encountered 904 unique sequences from 2914 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D143_F_filt.fastq.gz## Encountered 939 unique sequences from 2941 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D144_F_filt.fastq.gz## Encountered 1267 unique sequences from 4312 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D145_F_filt.fastq.gz## Encountered 1756 unique sequences from 6741 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D146_F_filt.fastq.gz## Encountered 1438 unique sequences from 4560 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D147_F_filt.fastq.gz## Encountered 3590 unique sequences from 15637 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D148_F_filt.fastq.gz## Encountered 2762 unique sequences from 11413 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D149_F_filt.fastq.gz## Encountered 3021 unique sequences from 12017 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D150_F_filt.fastq.gz## Encountered 1566 unique sequences from 5032 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D2_F_filt.fastq.gz## Encountered 3707 unique sequences from 18075 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D3_F_filt.fastq.gz## Encountered 1479 unique sequences from 6250 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D5_F_filt.fastq.gz## Encountered 1195 unique sequences from 4052 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D6_F_filt.fastq.gz## Encountered 1832 unique sequences from 7369 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D7_F_filt.fastq.gz## Encountered 1183 unique sequences from 4765 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D8_F_filt.fastq.gz## Encountered 1382 unique sequences from 4871 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D9_F_filt.fastq.gz## Encountered 1709 unique sequences from 6504 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/Mock_F_filt.fastq.gz## Encountered 897 unique sequences from 4314 total sequences read.derepRs <- derepFastq(filtRs, verbose=TRUE)## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D0_R_file.fastq.gz## Encountered 1660 unique sequences from 7113 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D1_R_file.fastq.gz## Encountered 1349 unique sequences from 5299 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D141_R_file.fastq.gz## Encountered 1335 unique sequences from 5463 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D142_R_file.fastq.gz## Encountered 853 unique sequences from 2914 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D143_R_file.fastq.gz## Encountered 880 unique sequences from 2941 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D144_R_file.fastq.gz## Encountered 1286 unique sequences from 4312 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D145_R_file.fastq.gz## Encountered 1803 unique sequences from 6741 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D146_R_file.fastq.gz## Encountered 1265 unique sequences from 4560 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D147_R_file.fastq.gz## Encountered 3414 unique sequences from 15637 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D148_R_file.fastq.gz## Encountered 2522 unique sequences from 11413 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D149_R_file.fastq.gz## Encountered 2771 unique sequences from 12017 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D150_R_file.fastq.gz## Encountered 1415 unique sequences from 5032 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D2_R_file.fastq.gz## Encountered 3290 unique sequences from 18075 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D3_R_file.fastq.gz## Encountered 1390 unique sequences from 6250 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D5_R_file.fastq.gz## Encountered 1134 unique sequences from 4052 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D6_R_file.fastq.gz## Encountered 1635 unique sequences from 7369 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D7_R_file.fastq.gz## Encountered 1084 unique sequences from 4765 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D8_R_file.fastq.gz## Encountered 1161 unique sequences from 4871 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/F3D9_R_file.fastq.gz## Encountered 1502 unique sequences from 6504 total sequences read.## Dereplicating sequence entries in Fastq file: ~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/filtered/Mock_R_file.fastq.gz## Encountered 732 unique sequences from 4314 total sequences read.# Name the derep-class objects by the sample names
names(derepFs) <- sample.names
names(derepRs) <- sample.namesIf using your own data
If you have a a big dataset, get the initial error rate estimates from a subsample of your data.
Sample Inference
We are now ready to infer the sequence variants in each sample (second dada pass)
# First with the Forward reads
dadaFs <- dada(derepFs, err = errF, multithread = TRUE)## Sample 1 - 7113 reads in 1979 unique sequences.
## Sample 2 - 5299 reads in 1639 unique sequences.
## Sample 3 - 5463 reads in 1477 unique sequences.
## Sample 4 - 2914 reads in 904 unique sequences.
## Sample 5 - 2941 reads in 939 unique sequences.
## Sample 6 - 4312 reads in 1267 unique sequences.
## Sample 7 - 6741 reads in 1756 unique sequences.
## Sample 8 - 4560 reads in 1438 unique sequences.
## Sample 9 - 15637 reads in 3590 unique sequences.
## Sample 10 - 11413 reads in 2762 unique sequences.
## Sample 11 - 12017 reads in 3021 unique sequences.
## Sample 12 - 5032 reads in 1566 unique sequences.
## Sample 13 - 18075 reads in 3707 unique sequences.
## Sample 14 - 6250 reads in 1479 unique sequences.
## Sample 15 - 4052 reads in 1195 unique sequences.
## Sample 16 - 7369 reads in 1832 unique sequences.
## Sample 17 - 4765 reads in 1183 unique sequences.
## Sample 18 - 4871 reads in 1382 unique sequences.
## Sample 19 - 6504 reads in 1709 unique sequences.
## Sample 20 - 4314 reads in 897 unique sequences.# Then with the Reverse reads
dadaRs <- dada(derepRs, err = errR, multithread = TRUE)## Sample 1 - 7113 reads in 1660 unique sequences.
## Sample 2 - 5299 reads in 1349 unique sequences.
## Sample 3 - 5463 reads in 1335 unique sequences.
## Sample 4 - 2914 reads in 853 unique sequences.
## Sample 5 - 2941 reads in 880 unique sequences.
## Sample 6 - 4312 reads in 1286 unique sequences.
## Sample 7 - 6741 reads in 1803 unique sequences.
## Sample 8 - 4560 reads in 1265 unique sequences.
## Sample 9 - 15637 reads in 3414 unique sequences.
## Sample 10 - 11413 reads in 2522 unique sequences.
## Sample 11 - 12017 reads in 2771 unique sequences.
## Sample 12 - 5032 reads in 1415 unique sequences.
## Sample 13 - 18075 reads in 3290 unique sequences.
## Sample 14 - 6250 reads in 1390 unique sequences.
## Sample 15 - 4052 reads in 1134 unique sequences.
## Sample 16 - 7369 reads in 1635 unique sequences.
## Sample 17 - 4765 reads in 1084 unique sequences.
## Sample 18 - 4871 reads in 1161 unique sequences.
## Sample 19 - 6504 reads in 1502 unique sequences.
## Sample 20 - 4314 reads in 732 unique sequences.# Inspect the dada-class objects returned by the dada function
dadaFs[[1]]## dada-class: object describing DADA2 denoising results
## 128 sample sequences were inferred from 1979 input unique sequences.
## Key parameters: OMEGA_A = 1e-40, BAND_SIZE = 16, USE_QUALS = TRUEdadaRs[[1]]## dada-class: object describing DADA2 denoising results
## 116 sample sequences were inferred from 1660 input unique sequences.
## Key parameters: OMEGA_A = 1e-40, BAND_SIZE = 16, USE_QUALS = TRUEWe can see that the algorithm has inferred 128 unique sequence variants from the forward reads and 116 from the reverse reads.
Merge Paired Reads
We can eliminate further spurious sequence variants by merging overlapping reads. The core function is mergePairs and it depends on the forward and reverse reads being in matching order at the time they were dereplicated.
# Merge the denoised forward and reverse reads
mergers <- mergePairs(dadaFs, derepFs, dadaRs, derepRs, verbose = TRUE )## 6600 paired-reads (in 105 unique pairings) successfully merged out of 7113 (in 254 pairings) input.## 5078 paired-reads (in 100 unique pairings) successfully merged out of 5299 (in 192 pairings) input.## 5047 paired-reads (in 78 unique pairings) successfully merged out of 5463 (in 199 pairings) input.## 2663 paired-reads (in 52 unique pairings) successfully merged out of 2914 (in 142 pairings) input.## 2575 paired-reads (in 54 unique pairings) successfully merged out of 2941 (in 162 pairings) input.## 3668 paired-reads (in 53 unique pairings) successfully merged out of 4312 (in 203 pairings) input.## 6203 paired-reads (in 81 unique pairings) successfully merged out of 6741 (in 230 pairings) input.## 4040 paired-reads (in 90 unique pairings) successfully merged out of 4560 (in 233 pairings) input.## 14340 paired-reads (in 142 unique pairings) successfully merged out of 15637 (in 409 pairings) input.## 10599 paired-reads (in 117 unique pairings) successfully merged out of 11413 (in 331 pairings) input.## 11196 paired-reads (in 134 unique pairings) successfully merged out of 12017 (in 343 pairings) input.## 4390 paired-reads (in 82 unique pairings) successfully merged out of 5032 (in 233 pairings) input.## 17477 paired-reads (in 148 unique pairings) successfully merged out of 18075 (in 329 pairings) input.## 5907 paired-reads (in 80 unique pairings) successfully merged out of 6250 (in 193 pairings) input.## 3770 paired-reads (in 85 unique pairings) successfully merged out of 4052 (in 192 pairings) input.## 6914 paired-reads (in 98 unique pairings) successfully merged out of 7369 (in 227 pairings) input.## 4480 paired-reads (in 66 unique pairings) successfully merged out of 4765 (in 159 pairings) input.## 4603 paired-reads (in 95 unique pairings) successfully merged out of 4871 (in 204 pairings) input.## 6173 paired-reads (in 108 unique pairings) successfully merged out of 6504 (in 196 pairings) input.## 4280 paired-reads (in 20 unique pairings) successfully merged out of 4314 (in 32 pairings) input.# Inspect the merged data.frame from the first sample
head(mergers[[1]])## sequence
## 1 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGCAGGCGGAAGATCAAGTCAGCGGTAAAATTGAGAGGCTCAACCTCTTCGAGCCGTTGAAACTGGTTTTCTTGAGTGAGCGAGAAGTATGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACTCCGATTGCGAAGGCAGCATACCGGCGCTCAACTGACGCTCATGCACGAAAGTGTGGGTATCGAACAGG
## 2 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGCCTGCCAAGTCAGCGGTAAAATTGCGGGGCTCAACCCCGTACAGCCGTTGAAACTGCCGGGCTCGAGTGGGCGAGAAGTATGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACCCCGATTGCGAAGGCAGCATACCGGCGCCCTACTGACGCTGAGGCACGAAAGTGCGGGGATCAAACAGG
## 3 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGGCTGTTAAGTCAGCGGTCAAATGTCGGGGCTCAACCCCGGCCTGCCGTTGAAACTGGCGGCCTCGAGTGGGCGAGAAGTATGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACTCCGATTGCGAAGGCAGCATACCGGCGCCCGACTGACGCTGAGGCACGAAAGCGTGGGTATCGAACAGG
## 4 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGGCTTTTAAGTCAGCGGTAAAAATTCGGGGCTCAACCCCGTCCGGCCGTTGAAACTGGGGGCCTTGAGTGGGCGAGAAGAAGGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACCCCGATTGCGAAGGCAGCCTTCCGGCGCCCTACTGACGCTGAGGCACGAAAGTGCGGGGATCGAACAGG
## 5 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGCAGGCGGACTCTCAAGTCAGCGGTCAAATCGCGGGGCTCAACCCCGTTCCGCCGTTGAAACTGGGAGCCTTGAGTGCGCGAGAAGTAGGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACTCCGATTGCGAAGGCAGCCTACCGGCGCGCAACTGACGCTCATGCACGAAAGCGTGGGTATCGAACAGG
## 6 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGGATGCCAAGTCAGCGGTAAAAAAGCGGTGCTCAACGCCGTCGAGCCGTTGAAACTGGCGTTCTTGAGTGGGCGAGAAGTATGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACTCCGATTGCGAAGGCAGCATACCGGCGCCCTACTGACGCTGAGGCACGAAAGCGTGGGTATCGAACAGG
## abundance forward reverse nmatch nmismatch nindel prefer accept
## 1 586 1 1 148 0 0 1 TRUE
## 2 471 2 2 148 0 0 2 TRUE
## 3 451 3 4 148 0 0 1 TRUE
## 4 433 4 3 148 0 0 2 TRUE
## 5 353 5 6 148 0 0 1 TRUE
## 6 285 6 5 148 0 0 2 TRUEWe now have a data.frame object for each sample with the merged $sequence, its abundance, and the indices of the merged $forward and $reverse denoised sequences. Pair reads that did not precisely overlap have been removed by the mergePairs function.
Important Note 5
If doing this with your own data, most of your reads should successfully merge. If this is not the case, you will need to revisit some upstream parameters. In particular, make sure you did not trim away any overlap between reads.
End of Note
Sequence Table Construction
We will now construct the sequence table, this being analogous to the “OTU table” produced by other methods.
# Construct sequence table
seqtab <- makeSequenceTable(mergers)## The sequences being tabled vary in length.# Consider the table
dim(seqtab)## [1] 20 288class(seqtab)## [1] "matrix"# Inspect the distribution of sequence lengths
table(nchar(getSequences(seqtab)))##
## 251 252 253 254 255
## 1 87 192 6 2We see that the sequence table is a matrix with rows corresponding to and named by the samples, and columns corresponding to (and named by) the sequence variants. We also see that the lengths of all of the sequences fall in the range expected for V4 amplicons.
Important Note 6
If working with your own data you may find sequences that are much longer or much shorter than expected. These may be the result of non-specific priming, and you should consider removing them. Use the command seqtab2 <- seqtab[,nchar(colnames(seqtab)) %in% seq(250, 256)].
End of Note
Remove Chimeras
So far we have let the dada function remove substitution errors and indel errors, but chimeras remain. The accuracy of the sequences after denoising makes chimera identification easier than if we had done that earlier with “fuzzier” sequences because all sequences now can be exactly reconstructed as a bimera (two-parent chimera) from more abundant sequences.
# Remove chimeric sequences
seqtab.nochim <- removeBimeraDenovo(seqtab, method = "consensus", multithread = TRUE, verbose=TRUE)## Identified 59 bimeras out of 288 input sequences.dim(seqtab.nochim)## [1] 20 229sum(seqtab.nochim)/sum(seqtab)## [1] 0.9643085The fraction of chimeras can be substantial. In this example, chimeras account for 59/288 unique sequence variants, or about 20% of them, but these variants account for only about 4% of the total sequence reads.
Important Note 7
Most of the reads should remain after chimera removal, although it is not uncommon for a majority of sequence variants to be removed. If most of your reads are removed as chimeric, you may need to revisit upstream processing. In almost all cases this is caused by primer sequences with ambiguous nucleotides that were not removed prior to beginning the DADA2 pipeline.
End of Note
Track Reads through the Pipeline
getN <- function(x) sum(getUniques(x))
pctSurv <- rowSums(seqtab.nochim)*100/out[,1]
track <- cbind(out, sapply(dadaFs, getN), sapply(mergers, getN), rowSums(seqtab), rowSums(seqtab.nochim), pctSurv)
colnames(track) <- c("input", "filtered", "denoised", "merged", "tabled", "nonchimeric", "% passing")
rownames(track) <- sample.names
head(track)## input filtered denoised merged tabled nonchimeric % passing
## F3D0 7793 7113 7113 6600 6600 6588 84.53741
## F3D1 5869 5299 5299 5078 5078 5067 86.33498
## F3D141 5958 5463 5463 5047 5047 4928 82.71232
## F3D142 3183 2914 2914 2663 2663 2600 81.68395
## F3D143 3178 2941 2941 2575 2575 2550 80.23914
## F3D144 4827 4312 4312 3668 3668 3527 73.06816Assign Taxonomy
Most people want to know the names of the organisms associated with the sequence variants, and so we want to classify them taxonomically. The package will use a classifier for this purpose, taking a set of sequences and a training set of taxonomically classified sequences, and outputs taxonomic assignments with at least minBoot bootstrap confidence.
There are many training sets to use. GreenGenes is one such set, but it has not been updated in 3 years. UNITED ITS and the Ribosomal Database Project (RDP) are others, the former being used for fungi. We are going to use a training set from SILVA. You should have downloaded that earlier and it should be sitting in the same folder as the original forward and reverse read files.
# Assign taxonomy
# First initialize random number generator for reproducibility
set.seed(100)
getwd()## [1] "/Users/vicki/Documents/NRSG_741/2018/2018Week7"path## [1] "~/Documents/NRSG_741/MiSeqData/MiSeq_SOP"list.files(path)## [1] "F3D0_S188_L001_R1_001.fastq"
## [2] "F3D0_S188_L001_R2_001.fastq"
## [3] "F3D1_S189_L001_R1_001.fastq"
## [4] "F3D1_S189_L001_R2_001.fastq"
## [5] "F3D141_S207_L001_R1_001.fastq"
## [6] "F3D141_S207_L001_R2_001.fastq"
## [7] "F3D142_S208_L001_R1_001.fastq"
## [8] "F3D142_S208_L001_R2_001.fastq"
## [9] "F3D143_S209_L001_R1_001.fastq"
## [10] "F3D143_S209_L001_R2_001.fastq"
## [11] "F3D144_S210_L001_R1_001.fastq"
## [12] "F3D144_S210_L001_R2_001.fastq"
## [13] "F3D145_S211_L001_R1_001.fastq"
## [14] "F3D145_S211_L001_R2_001.fastq"
## [15] "F3D146_S212_L001_R1_001.fastq"
## [16] "F3D146_S212_L001_R2_001.fastq"
## [17] "F3D147_S213_L001_R1_001.fastq"
## [18] "F3D147_S213_L001_R2_001.fastq"
## [19] "F3D148_S214_L001_R1_001.fastq"
## [20] "F3D148_S214_L001_R2_001.fastq"
## [21] "F3D149_S215_L001_R1_001.fastq"
## [22] "F3D149_S215_L001_R2_001.fastq"
## [23] "F3D150_S216_L001_R1_001.fastq"
## [24] "F3D150_S216_L001_R2_001.fastq"
## [25] "F3D2_S190_L001_R1_001.fastq"
## [26] "F3D2_S190_L001_R2_001.fastq"
## [27] "F3D3_S191_L001_R1_001.fastq"
## [28] "F3D3_S191_L001_R2_001.fastq"
## [29] "F3D5_S193_L001_R1_001.fastq"
## [30] "F3D5_S193_L001_R2_001.fastq"
## [31] "F3D6_S194_L001_R1_001.fastq"
## [32] "F3D6_S194_L001_R2_001.fastq"
## [33] "F3D7_S195_L001_R1_001.fastq"
## [34] "F3D7_S195_L001_R2_001.fastq"
## [35] "F3D8_S196_L001_R1_001.fastq"
## [36] "F3D8_S196_L001_R2_001.fastq"
## [37] "F3D9_S197_L001_R1_001.fastq"
## [38] "F3D9_S197_L001_R2_001.fastq"
## [39] "filtered"
## [40] "HMP_MOCK.v35.fasta"
## [41] "Mock_S280_L001_R1_001.fastq"
## [42] "Mock_S280_L001_R2_001.fastq"
## [43] "mouse.dpw.metadata"
## [44] "mouse.time.design"
## [45] "raw quality plots"
## [46] "rdp_species_assignment_14.fa.gz"
## [47] "rdp_train_set_14.fa"
## [48] "rdp_train_set_14.fa.gz"
## [49] "silva_nr_v132_train_set.fa.gz"
## [50] "silva_species_assignment_v132.fa.gz"
## [51] "stability.batch"
## [52] "stability.files"taxa <- assignTaxonomy(seqtab.nochim, "~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/silva_nr_v132_train_set.fa.gz", multithread = TRUE)
unname(head(taxa))## [,1] [,2] [,3] [,4]
## [1,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales"
## [2,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales"
## [3,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales"
## [4,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales"
## [5,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales"
## [6,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales"
## [,5] [,6]
## [1,] "Muribaculaceae" NA
## [2,] "Muribaculaceae" NA
## [3,] "Muribaculaceae" NA
## [4,] "Muribaculaceae" NA
## [5,] "Bacteroidaceae" "Bacteroides"
## [6,] "Muribaculaceae" NASpecies Assignment
We can also use the SILVA species assignment dataset to do exactly that, that is, to assign species.
# Assign species
taxa <- addSpecies(taxa, "~/Documents/NRSG_741/MiSeqData/MiSeq_SOP/silva_species_assignment_v132.fa.gz")Inspect the taxonomic assignments:
taxa.print <- taxa #Removing sequence rownames for display only
rownames (taxa.print) <- NULL
head(taxa.print)## Kingdom Phylum Class Order
## [1,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales"
## [2,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales"
## [3,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales"
## [4,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales"
## [5,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales"
## [6,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales"
## Family Genus Species
## [1,] "Muribaculaceae" NA NA
## [2,] "Muribaculaceae" NA NA
## [3,] "Muribaculaceae" NA NA
## [4,] "Muribaculaceae" NA NA
## [5,] "Bacteroidaceae" "Bacteroides" NA
## [6,] "Muribaculaceae" NA NAEvaluate Accuracy
In addition to the MiSeq_SOP files, we have also analyzed a “mock community”, a mixture of 20 known strains. Reference sequences were provided in the downloaded zip archive. Let’s see how the sequences inferred by DADA2 compare to the expected composition of the community.
# Evaluate DADA2's accuracy on the mock community
unqs.mock <- seqtab.nochim["Mock",]
unqs.mock <- sort(unqs.mock[unqs.mock>0], decreasing=TRUE) #Drop ASVs absent in the Mock Community
cat("DADA2 inferred", length(unqs.mock), "sample sequences present in the Mock Community. \n")## DADA2 inferred 20 sample sequences present in the Mock Community.mock.ref <- getSequences(file.path(path, "HMP_MOCK.v35.fasta"))
match.ref <- sum(sapply(names(unqs.mock), function(x)any(grepl(x, mock.ref))))
cat("Of those,", sum(match.ref), "were exact matches to the expected reference sequences. \n")## Of those, 20 were exact matches to the expected reference sequences.Construct a Phylogenetic Tree
Phylogenetic relatedness is often used to inform downstream analyses, particularly the calculation of phylogeny-aware distances between microbial communities, including the weighted and unweighted UniFrac distances. We can use the reference-free dada2 sequence inference to construct the phylogenetic tree by relating the inferred sequence variants de novo. First we will need to install the DECIPHER package from Bioconductor and the phangorn package from CRAN.
Next call them up then we will use DECIPHER for multiple sequence alignment
library(DECIPHER)## Loading required package: RSQLiteseqs <- getSequences(seqtab.nochim)
# This next command will allow propagation of sequence names to the tip labels of the tree
names(seqs) <- seqs
alignment <- AlignSeqs(DNAStringSet(seqs), anchor=NA)## Determining distance matrix based on shared 7-mers:
##
|
| | 0%
|
|= | 1%
|
|= | 2%
|
|== | 3%
|
|=== | 4%
|
|=== | 5%
|
|==== | 6%
|
|===== | 7%
|
|===== | 8%
|
|====== | 9%
|
|====== | 10%
|
|======= | 11%
|
|======== | 12%
|
|======== | 13%
|
|========= | 14%
|
|========== | 15%
|
|========== | 16%
|
|=========== | 17%
|
|============ | 18%
|
|============ | 19%
|
|============= | 20%
|
|============== | 21%
|
|============== | 22%
|
|=============== | 23%
|
|================ | 24%
|
|================ | 25%
|
|================= | 26%
|
|================== | 27%
|
|================== | 28%
|
|=================== | 29%
|
|==================== | 30%
|
|==================== | 31%
|
|===================== | 32%
|
|===================== | 33%
|
|====================== | 34%
|
|======================= | 35%
|
|======================= | 36%
|
|======================== | 37%
|
|========================= | 38%
|
|========================= | 39%
|
|========================== | 40%
|
|=========================== | 41%
|
|=========================== | 42%
|
|============================ | 43%
|
|============================= | 44%
|
|============================= | 45%
|
|============================== | 46%
|
|=============================== | 47%
|
|=============================== | 48%
|
|================================ | 49%
|
|================================ | 50%
|
|================================= | 51%
|
|================================== | 52%
|
|================================== | 53%
|
|=================================== | 54%
|
|==================================== | 55%
|
|==================================== | 56%
|
|===================================== | 57%
|
|====================================== | 58%
|
|====================================== | 59%
|
|======================================= | 60%
|
|======================================== | 61%
|
|======================================== | 62%
|
|========================================= | 63%
|
|========================================== | 64%
|
|========================================== | 65%
|
|=========================================== | 66%
|
|============================================ | 67%
|
|============================================ | 68%
|
|============================================= | 69%
|
|============================================== | 70%
|
|============================================== | 71%
|
|=============================================== | 72%
|
|=============================================== | 73%
|
|================================================ | 74%
|
|================================================= | 75%
|
|================================================= | 76%
|
|================================================== | 77%
|
|=================================================== | 78%
|
|=================================================== | 79%
|
|==================================================== | 80%
|
|===================================================== | 81%
|
|===================================================== | 82%
|
|====================================================== | 83%
|
|======================================================= | 84%
|
|======================================================= | 85%
|
|======================================================== | 86%
|
|========================================================= | 87%
|
|========================================================= | 88%
|
|========================================================== | 89%
|
|========================================================== | 90%
|
|=========================================================== | 91%
|
|============================================================ | 92%
|
|============================================================ | 93%
|
|============================================================= | 94%
|
|============================================================== | 95%
|
|============================================================== | 96%
|
|=============================================================== | 97%
|
|================================================================ | 98%
|
|================================================================ | 99%
|
|=================================================================| 100%
##
## Time difference of 0.33 secs
##
## Clustering into groups by similarity:
##
|
| | 0%
|
|= | 1%
|
|= | 2%
|
|== | 3%
|
|=== | 4%
|
|=== | 5%
|
|==== | 6%
|
|===== | 7%
|
|===== | 8%
|
|====== | 9%
|
|====== | 10%
|
|======= | 11%
|
|======== | 12%
|
|======== | 13%
|
|========= | 14%
|
|========== | 15%
|
|========== | 16%
|
|=========== | 17%
|
|============ | 18%
|
|============ | 19%
|
|============= | 20%
|
|============== | 21%
|
|============== | 22%
|
|=============== | 23%
|
|================ | 24%
|
|================ | 25%
|
|================= | 26%
|
|================== | 27%
|
|================== | 28%
|
|=================== | 29%
|
|==================== | 30%
|
|==================== | 31%
|
|===================== | 32%
|
|===================== | 33%
|
|====================== | 34%
|
|======================= | 35%
|
|======================= | 36%
|
|======================== | 37%
|
|========================= | 38%
|
|========================= | 39%
|
|========================== | 40%
|
|=========================== | 41%
|
|=========================== | 42%
|
|============================ | 43%
|
|============================= | 44%
|
|============================= | 45%
|
|============================== | 46%
|
|=============================== | 47%
|
|=============================== | 48%
|
|================================ | 49%
|
|================================ | 50%
|
|================================= | 51%
|
|================================== | 52%
|
|================================== | 53%
|
|=================================== | 54%
|
|==================================== | 55%
|
|==================================== | 56%
|
|===================================== | 57%
|
|====================================== | 58%
|
|====================================== | 59%
|
|======================================= | 60%
|
|======================================== | 61%
|
|======================================== | 62%
|
|========================================= | 63%
|
|========================================== | 64%
|
|========================================== | 65%
|
|=========================================== | 66%
|
|============================================ | 67%
|
|============================================ | 68%
|
|============================================= | 69%
|
|============================================== | 70%
|
|============================================== | 71%
|
|=============================================== | 72%
|
|=============================================== | 73%
|
|================================================ | 74%
|
|================================================= | 75%
|
|================================================= | 76%
|
|================================================== | 77%
|
|=================================================== | 78%
|
|=================================================== | 79%
|
|==================================================== | 80%
|
|===================================================== | 81%
|
|===================================================== | 82%
|
|====================================================== | 83%
|
|======================================================= | 84%
|
|======================================================= | 85%
|
|======================================================== | 86%
|
|========================================================= | 87%
|
|========================================================= | 88%
|
|========================================================== | 89%
|
|========================================================== | 90%
|
|=========================================================== | 91%
|
|============================================================ | 92%
|
|============================================================ | 93%
|
|============================================================= | 94%
|
|============================================================== | 95%
|
|============================================================== | 96%
|
|=============================================================== | 97%
|
|================================================================ | 98%
|
|================================================================ | 99%
|
|=================================================================| 100%
##
## Time difference of 0.12 secs
##
## Aligning Sequences:
##
|
| | 0%
|
|= | 1%
|
|= | 2%
|
|== | 3%
|
|=== | 4%
|
|=== | 5%
|
|==== | 6%
|
|===== | 7%
|
|===== | 8%
|
|====== | 9%
|
|====== | 10%
|
|======= | 11%
|
|======== | 12%
|
|======== | 13%
|
|========= | 14%
|
|========== | 15%
|
|========== | 16%
|
|=========== | 17%
|
|============ | 18%
|
|============ | 19%
|
|============= | 20%
|
|============== | 21%
|
|============== | 22%
|
|=============== | 23%
|
|================ | 24%
|
|================ | 25%
|
|================= | 26%
|
|================== | 27%
|
|================== | 28%
|
|=================== | 29%
|
|==================== | 30%
|
|==================== | 31%
|
|===================== | 32%
|
|===================== | 33%
|
|====================== | 34%
|
|======================= | 35%
|
|======================= | 36%
|
|======================== | 37%
|
|========================= | 38%
|
|========================= | 39%
|
|========================== | 40%
|
|=========================== | 41%
|
|=========================== | 42%
|
|============================ | 43%
|
|============================= | 44%
|
|============================= | 45%
|
|============================== | 46%
|
|=============================== | 47%
|
|=============================== | 48%
|
|================================ | 49%
|
|================================ | 50%
|
|================================= | 51%
|
|================================== | 52%
|
|================================== | 53%
|
|=================================== | 54%
|
|==================================== | 55%
|
|==================================== | 56%
|
|===================================== | 57%
|
|====================================== | 58%
|
|====================================== | 59%
|
|======================================= | 60%
|
|======================================== | 61%
|
|======================================== | 62%
|
|========================================= | 63%
|
|========================================== | 64%
|
|========================================== | 65%
|
|=========================================== | 66%
|
|============================================ | 67%
|
|============================================ | 68%
|
|============================================= | 69%
|
|============================================== | 70%
|
|============================================== | 71%
|
|=============================================== | 72%
|
|=============================================== | 73%
|
|================================================ | 74%
|
|================================================= | 75%
|
|================================================= | 76%
|
|================================================== | 77%
|
|=================================================== | 78%
|
|=================================================== | 79%
|
|==================================================== | 80%
|
|===================================================== | 81%
|
|===================================================== | 82%
|
|====================================================== | 83%
|
|======================================================= | 84%
|
|======================================================= | 85%
|
|======================================================== | 86%
|
|========================================================= | 87%
|
|========================================================= | 88%
|
|========================================================== | 89%
|
|========================================================== | 90%
|
|=========================================================== | 91%
|
|============================================================ | 92%
|
|============================================================ | 93%
|
|============================================================= | 94%
|
|============================================================== | 95%
|
|============================================================== | 96%
|
|=============================================================== | 97%
|
|================================================================ | 98%
|
|================================================================ | 99%
|
|=================================================================| 100%
##
## Time difference of 1.33 secs
##
## Iteration 1 of 2:
##
## Determining distance matrix based on alignment:
##
|
| | 0%
|
|= | 1%
|
|= | 2%
|
|== | 3%
|
|=== | 4%
|
|=== | 5%
|
|==== | 6%
|
|===== | 7%
|
|===== | 8%
|
|====== | 9%
|
|====== | 10%
|
|======= | 11%
|
|======== | 12%
|
|======== | 13%
|
|========= | 14%
|
|========== | 15%
|
|========== | 16%
|
|=========== | 17%
|
|============ | 18%
|
|============ | 19%
|
|============= | 20%
|
|============== | 21%
|
|============== | 22%
|
|=============== | 23%
|
|================ | 24%
|
|================ | 25%
|
|================= | 26%
|
|================== | 27%
|
|================== | 28%
|
|=================== | 29%
|
|==================== | 30%
|
|==================== | 31%
|
|===================== | 32%
|
|===================== | 33%
|
|====================== | 34%
|
|======================= | 35%
|
|======================= | 36%
|
|======================== | 37%
|
|========================= | 38%
|
|========================= | 39%
|
|========================== | 40%
|
|=========================== | 41%
|
|=========================== | 42%
|
|============================ | 43%
|
|============================= | 44%
|
|============================= | 45%
|
|============================== | 46%
|
|=============================== | 47%
|
|=============================== | 48%
|
|================================ | 49%
|
|================================ | 50%
|
|================================= | 51%
|
|================================== | 52%
|
|================================== | 53%
|
|=================================== | 54%
|
|==================================== | 55%
|
|==================================== | 56%
|
|===================================== | 57%
|
|====================================== | 58%
|
|====================================== | 59%
|
|======================================= | 60%
|
|======================================== | 61%
|
|======================================== | 62%
|
|========================================= | 63%
|
|========================================== | 64%
|
|========================================== | 65%
|
|=========================================== | 66%
|
|============================================ | 67%
|
|============================================ | 68%
|
|============================================= | 69%
|
|============================================== | 70%
|
|============================================== | 71%
|
|=============================================== | 72%
|
|=============================================== | 73%
|
|================================================ | 74%
|
|================================================= | 75%
|
|================================================= | 76%
|
|================================================== | 77%
|
|=================================================== | 78%
|
|=================================================== | 79%
|
|==================================================== | 80%
|
|===================================================== | 81%
|
|===================================================== | 82%
|
|====================================================== | 83%
|
|======================================================= | 84%
|
|======================================================= | 85%
|
|======================================================== | 86%
|
|========================================================= | 87%
|
|========================================================= | 88%
|
|========================================================== | 89%
|
|========================================================== | 90%
|
|=========================================================== | 91%
|
|============================================================ | 92%
|
|============================================================ | 93%
|
|============================================================= | 94%
|
|============================================================== | 95%
|
|============================================================== | 96%
|
|=============================================================== | 97%
|
|================================================================ | 98%
|
|================================================================ | 99%
|
|=================================================================| 100%
##
## Time difference of 0.03 secs
##
## Reclustering into groups by similarity:
##
|
| | 0%
|
|= | 1%
|
|= | 2%
|
|== | 3%
|
|=== | 4%
|
|=== | 5%
|
|==== | 6%
|
|===== | 7%
|
|===== | 8%
|
|====== | 9%
|
|====== | 10%
|
|======= | 11%
|
|======== | 12%
|
|======== | 13%
|
|========= | 14%
|
|========== | 15%
|
|========== | 16%
|
|=========== | 17%
|
|============ | 18%
|
|============ | 19%
|
|============= | 20%
|
|============== | 21%
|
|============== | 22%
|
|=============== | 23%
|
|================ | 24%
|
|================ | 25%
|
|================= | 26%
|
|================== | 27%
|
|================== | 28%
|
|=================== | 29%
|
|==================== | 30%
|
|==================== | 31%
|
|===================== | 32%
|
|===================== | 33%
|
|====================== | 34%
|
|======================= | 35%
|
|======================= | 36%
|
|======================== | 37%
|
|========================= | 38%
|
|========================= | 39%
|
|========================== | 40%
|
|=========================== | 41%
|
|=========================== | 42%
|
|============================ | 43%
|
|============================= | 44%
|
|============================= | 45%
|
|============================== | 46%
|
|=============================== | 47%
|
|=============================== | 48%
|
|================================ | 49%
|
|================================ | 50%
|
|================================= | 51%
|
|================================== | 52%
|
|================================== | 53%
|
|=================================== | 54%
|
|==================================== | 55%
|
|==================================== | 56%
|
|===================================== | 57%
|
|====================================== | 58%
|
|====================================== | 59%
|
|======================================= | 60%
|
|======================================== | 61%
|
|======================================== | 62%
|
|========================================= | 63%
|
|========================================== | 64%
|
|========================================== | 65%
|
|=========================================== | 66%
|
|============================================ | 67%
|
|============================================ | 68%
|
|============================================= | 69%
|
|============================================== | 70%
|
|============================================== | 71%
|
|=============================================== | 72%
|
|=============================================== | 73%
|
|================================================ | 74%
|
|================================================= | 75%
|
|================================================= | 76%
|
|================================================== | 77%
|
|=================================================== | 78%
|
|=================================================== | 79%
|
|==================================================== | 80%
|
|===================================================== | 81%
|
|===================================================== | 82%
|
|====================================================== | 83%
|
|======================================================= | 84%
|
|======================================================= | 85%
|
|======================================================== | 86%
|
|========================================================= | 87%
|
|========================================================= | 88%
|
|========================================================== | 89%
|
|========================================================== | 90%
|
|=========================================================== | 91%
|
|============================================================ | 92%
|
|============================================================ | 93%
|
|============================================================= | 94%
|
|============================================================== | 95%
|
|============================================================== | 96%
|
|=============================================================== | 97%
|
|================================================================ | 98%
|
|================================================================ | 99%
|
|=================================================================| 100%
##
## Time difference of 0.07 secs
##
## Realigning Sequences:
##
|
| | 0%
|
|= | 1%
|
|= | 2%
|
|== | 3%
|
|=== | 4%
|
|=== | 5%
|
|==== | 6%
|
|===== | 7%
|
|===== | 8%
|
|====== | 9%
|
|====== | 10%
|
|======= | 11%
|
|======== | 12%
|
|======== | 13%
|
|========= | 14%
|
|========== | 15%
|
|========== | 16%
|
|=========== | 17%
|
|============ | 18%
|
|============ | 19%
|
|============= | 20%
|
|============== | 21%
|
|============== | 22%
|
|=============== | 23%
|
|================ | 24%
|
|================ | 25%
|
|================= | 26%
|
|================== | 27%
|
|================== | 28%
|
|=================== | 29%
|
|==================== | 30%
|
|==================== | 31%
|
|===================== | 32%
|
|===================== | 33%
|
|====================== | 34%
|
|======================= | 35%
|
|======================= | 36%
|
|======================== | 37%
|
|========================= | 38%
|
|========================= | 39%
|
|========================== | 40%
|
|=========================== | 41%
|
|=========================== | 42%
|
|============================ | 43%
|
|============================= | 44%
|
|============================= | 45%
|
|============================== | 46%
|
|=============================== | 47%
|
|=============================== | 48%
|
|================================ | 49%
|
|================================ | 50%
|
|================================= | 51%
|
|================================== | 52%
|
|================================== | 53%
|
|=================================== | 54%
|
|==================================== | 55%
|
|==================================== | 56%
|
|===================================== | 57%
|
|====================================== | 58%
|
|====================================== | 59%
|
|======================================= | 60%
|
|======================================== | 61%
|
|======================================== | 62%
|
|========================================= | 63%
|
|========================================== | 64%
|
|========================================== | 65%
|
|=========================================== | 66%
|
|============================================ | 67%
|
|============================================ | 68%
|
|============================================= | 69%
|
|============================================== | 70%
|
|============================================== | 71%
|
|=============================================== | 72%
|
|=============================================== | 73%
|
|================================================ | 74%
|
|================================================= | 75%
|
|================================================= | 76%
|
|================================================== | 77%
|
|=================================================== | 78%
|
|=================================================== | 79%
|
|==================================================== | 80%
|
|===================================================== | 81%
|
|===================================================== | 82%
|
|====================================================== | 83%
|
|======================================================= | 84%
|
|======================================================= | 85%
|
|======================================================== | 86%
|
|========================================================= | 87%
|
|========================================================= | 88%
|
|========================================================== | 89%
|
|========================================================== | 90%
|
|=========================================================== | 91%
|
|============================================================ | 92%
|
|============================================================ | 93%
|
|============================================================= | 94%
|
|============================================================== | 95%
|
|============================================================== | 96%
|
|=============================================================== | 97%
|
|================================================================ | 98%
|
|================================================================ | 99%
|
|=================================================================| 100%
##
## Time difference of 0.82 secs
##
## Iteration 2 of 2:
##
## Determining distance matrix based on alignment:
##
|
| | 0%
|
|= | 1%
|
|= | 2%
|
|== | 3%
|
|=== | 4%
|
|=== | 5%
|
|==== | 6%
|
|===== | 7%
|
|===== | 8%
|
|====== | 9%
|
|====== | 10%
|
|======= | 11%
|
|======== | 12%
|
|======== | 13%
|
|========= | 14%
|
|========== | 15%
|
|========== | 16%
|
|=========== | 17%
|
|============ | 18%
|
|============ | 19%
|
|============= | 20%
|
|============== | 21%
|
|============== | 22%
|
|=============== | 23%
|
|================ | 24%
|
|================ | 25%
|
|================= | 26%
|
|================== | 27%
|
|================== | 28%
|
|=================== | 29%
|
|==================== | 30%
|
|==================== | 31%
|
|===================== | 32%
|
|===================== | 33%
|
|====================== | 34%
|
|======================= | 35%
|
|======================= | 36%
|
|======================== | 37%
|
|========================= | 38%
|
|========================= | 39%
|
|========================== | 40%
|
|=========================== | 41%
|
|=========================== | 42%
|
|============================ | 43%
|
|============================= | 44%
|
|============================= | 45%
|
|============================== | 46%
|
|=============================== | 47%
|
|=============================== | 48%
|
|================================ | 49%
|
|================================ | 50%
|
|================================= | 51%
|
|================================== | 52%
|
|================================== | 53%
|
|=================================== | 54%
|
|==================================== | 55%
|
|==================================== | 56%
|
|===================================== | 57%
|
|====================================== | 58%
|
|====================================== | 59%
|
|======================================= | 60%
|
|======================================== | 61%
|
|======================================== | 62%
|
|========================================= | 63%
|
|========================================== | 64%
|
|========================================== | 65%
|
|=========================================== | 66%
|
|============================================ | 67%
|
|============================================ | 68%
|
|============================================= | 69%
|
|============================================== | 70%
|
|============================================== | 71%
|
|=============================================== | 72%
|
|=============================================== | 73%
|
|================================================ | 74%
|
|================================================= | 75%
|
|================================================= | 76%
|
|================================================== | 77%
|
|=================================================== | 78%
|
|=================================================== | 79%
|
|==================================================== | 80%
|
|===================================================== | 81%
|
|===================================================== | 82%
|
|====================================================== | 83%
|
|======================================================= | 84%
|
|======================================================= | 85%
|
|======================================================== | 86%
|
|========================================================= | 87%
|
|========================================================= | 88%
|
|========================================================== | 89%
|
|========================================================== | 90%
|
|=========================================================== | 91%
|
|============================================================ | 92%
|
|============================================================ | 93%
|
|============================================================= | 94%
|
|============================================================== | 95%
|
|============================================================== | 96%
|
|=============================================================== | 97%
|
|================================================================ | 98%
|
|================================================================ | 99%
|
|=================================================================| 100%
##
## Time difference of 0.02 secs
##
## Reclustering into groups by similarity:
##
|
| | 0%
|
|= | 1%
|
|= | 2%
|
|== | 3%
|
|=== | 4%
|
|=== | 5%
|
|==== | 6%
|
|===== | 7%
|
|===== | 8%
|
|====== | 9%
|
|====== | 10%
|
|======= | 11%
|
|======== | 12%
|
|======== | 13%
|
|========= | 14%
|
|========== | 15%
|
|========== | 16%
|
|=========== | 17%
|
|============ | 18%
|
|============ | 19%
|
|============= | 20%
|
|============== | 21%
|
|============== | 22%
|
|=============== | 23%
|
|================ | 24%
|
|================ | 25%
|
|================= | 26%
|
|================== | 27%
|
|================== | 28%
|
|=================== | 29%
|
|==================== | 30%
|
|==================== | 31%
|
|===================== | 32%
|
|===================== | 33%
|
|====================== | 34%
|
|======================= | 35%
|
|======================= | 36%
|
|======================== | 37%
|
|========================= | 38%
|
|========================= | 39%
|
|========================== | 40%
|
|=========================== | 41%
|
|=========================== | 42%
|
|============================ | 43%
|
|============================= | 44%
|
|============================= | 45%
|
|============================== | 46%
|
|=============================== | 47%
|
|=============================== | 48%
|
|================================ | 49%
|
|================================ | 50%
|
|================================= | 51%
|
|================================== | 52%
|
|================================== | 53%
|
|=================================== | 54%
|
|==================================== | 55%
|
|==================================== | 56%
|
|===================================== | 57%
|
|====================================== | 58%
|
|====================================== | 59%
|
|======================================= | 60%
|
|======================================== | 61%
|
|======================================== | 62%
|
|========================================= | 63%
|
|========================================== | 64%
|
|========================================== | 65%
|
|=========================================== | 66%
|
|============================================ | 67%
|
|============================================ | 68%
|
|============================================= | 69%
|
|============================================== | 70%
|
|============================================== | 71%
|
|=============================================== | 72%
|
|=============================================== | 73%
|
|================================================ | 74%
|
|================================================= | 75%
|
|================================================= | 76%
|
|================================================== | 77%
|
|=================================================== | 78%
|
|=================================================== | 79%
|
|==================================================== | 80%
|
|===================================================== | 81%
|
|===================================================== | 82%
|
|====================================================== | 83%
|
|======================================================= | 84%
|
|======================================================= | 85%
|
|======================================================== | 86%
|
|========================================================= | 87%
|
|========================================================= | 88%
|
|========================================================== | 89%
|
|========================================================== | 90%
|
|=========================================================== | 91%
|
|============================================================ | 92%
|
|============================================================ | 93%
|
|============================================================= | 94%
|
|============================================================== | 95%
|
|============================================================== | 96%
|
|=============================================================== | 97%
|
|================================================================ | 98%
|
|================================================================ | 99%
|
|=================================================================| 100%
##
## Time difference of 0.09 secs
##
## Realigning Sequences:
##
|
| | 0%
|
|=== | 4%
|
|====== | 9%
|
|======== | 13%
|
|============ | 18%
|
|============== | 22%
|
|================== | 27%
|
|==================== | 31%
|
|======================= | 36%
|
|========================== | 40%
|
|============================= | 45%
|
|================================ | 50%
|
|=================================== | 54%
|
|====================================== | 59%
|
|========================================= | 63%
|
|============================================ | 68%
|
|=============================================== | 72%
|
|================================================== | 77%
|
|===================================================== | 81%
|
|======================================================== | 86%
|
|========================================================== | 90%
|
|============================================================== | 95%
|
|=================================================================| 100%
##
## Time difference of 0.16 secsNow that the sequences are aligned we can use the phanghorn package to construct the tree.
library(phangorn)## Loading required package: ape##
## Attaching package: 'ape'## The following object is masked from 'package:ShortRead':
##
## zoom## The following object is masked from 'package:Biostrings':
##
## complement# Construct the tree
phang.align <- phyDat(as(alignment, "matrix"), type="DNA")
dm <- dist.ml(phang.align)
treeNJ <- NJ(dm) # Tip order will not equal sequence order
fit <- pml(treeNJ, data=phang.align)## negative edges length changed to 0!## negative edges length changed to 0.
fitGTR <- update(fit, k=4, inv=0.2)
fitGTR <- optim.pml(fitGTR, model="GTR", optInv=TRUE, optGamma=TRUE,
rearrangement = "stochastic", control=pml.control(trace=0))
detach("package:phangorn", unload=TRUE)We will be viewing the tree below.
Prepare for Piphillin
We are now in a position where we can prepare our output for uploading to Piphillin http://www.secondgenome.com/solutions/resources/data-analysis-tools/piphillin/. Piphillin is a tool to infer function from 16S rRNA abundance. It takes the data (more ore less) directly from the sequence variant table that dada2 produces.
You have to upload two items, a .fasta formatted file containing the representative sequences, and the sequence variant table (reformatted) to the webpage above, and it should return results to you in about 20 minutes.
To do this we need to work with our dada2 output a little bit more to create the .
# create the fasta format file for piphillin
fastseq <- colnames(seqtab.nochim)
start <- as.character(seq(1:length(fastseq)))
otustart <- paste(">otu", start, sep="")
fsta <- c(rbind(otustart, fastseq))
write(fsta, "example.fasta")
# write out the transposed seqtab.nochim file in
# .csv format
otulab <- paste("otu", start, seq="")
stnct <-t(seqtab.nochim)
rownames(stnct) <- otulab
write.csv(stnct, "seqtab3.csv")
# use seqtab3.csv and example.fasta
# as the input files for piphillinOnce you have these two files exported, use a text editor to open the .csv file, and change the first line to start with “OTU,samplename1,samplename2,etc.”.
Then go to the website above, fill out the form, upload these files, and submit the request. Choose to get both KEGG and BIOCYC results from the latest available databases, and ask for 99% accuracy. You will get an emailed response in about 20 minutes with a link to your results. Download those results immediately (the link and results will disappear in 24 hours). Unzipe the file, and look in each of the _output folders. There should be some .txt files in both of them (if you have done everything correctly). Store these files somewhere on your computer and we will come back to these files in a later lecture.
Handoff to phyloseq
Our next activity will be to hand off the data to the phyloseq package for analysis. This package requires three items: the “OTUtable,” the taxonomy table, and data about the samples. The first two items are directly available at the end of your dada2run, and you can import the latter as a .csv file. In the case of the data that are considered here, we can calculate the derive the gender (G), mouse subject number (X), and day post-weaning (Y) directly from the file name, which has the form GXDY.
# Create a data frame for the sample data
samples.out <- rownames(seqtab.nochim)
# Create subject, gender, and day variables
subject <- sapply(strsplit(samples.out, "D"), `[`, 1)
gender <- substr(subject,1,1)
subject <- substr(subject, 2, 999)
day <- as.integer(sapply(strsplit(samples.out, "D"), `[`, 2))
# Combine into dataframe
samdf <- data.frame(Subject = subject, Gender = gender, Day = day)
#Create indicator of early or late day of post-weaning
samdf$When <- "Early"
samdf$When[samdf$Day > 100] <- "Late"
# Assign rownames to the dataframe == these will be the same as the rownames of the "OTUtable"
rownames(samdf) <- samples.outNow that we have our sample data, let’s create the phyloseq object.
library(phyloseq)
# Create phyloseq object
ps <- phyloseq(otu_table(seqtab.nochim, taxa_are_rows=FALSE),
sample_data(samdf),
tax_table(taxa),
phy_tree(fitGTR$tree))
# Describe it
ps## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 229 taxa and 20 samples ]
## sample_data() Sample Data: [ 20 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 229 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 229 tips and 227 internal nodes ]So we are now ready to use phyloseq. I will show you a few things you can do with these data. In our next session I will show you much much more.
Diversity in Microbial Ecology
A key concept in ecology in general, microbial ecology and microbiome research in particular is that of diversity. Often the term “species diversity” is used, although sometimes we do not have species level resolution to the species level. We can conceive of diversity at each taxonomic level, that is, genus diversity, family diversity, etc.
Whatever the level, the term \(\alpha\)-diversity is used to denote diversity in an individual setting. In microbiome studies, this typically means for each experimental unit measured, that is, person, animal, etc., the diversity within that experimental unit. Diversity at this level consists of two parts: richness and evenness.
- Richness: How many different types of units (e.g., species) are there?
- Evenness: How equal are the abundances of the different types?
Richness is a simple count.
There are several different measures of evenness. One common measure is the Shannon Index defined as
\[\begin{equation} H=-\sum_{i=1}^R p_i ln(p_i) \end{equation}\]Where R is the number of different types of units, and \(p_i\) is the proportion of units in type \(i\). When all types are equally common, then \(p_i=R, \all i\), and H = ln(R). If one type dominates at the expense of all others, then H –> 0. If there is only one type present, then H = 0.
Another common measure is the Simpson Index defined as
\[\begin{equation} \lambda = \sum_{i=1}^R p_i^2 \end{equation}\]When all types are equally abundant, then \(\lambda = 1/R\), and if one types dominates then \(\lambda\) –> 1.
Let’s see what these animals look like interms of individual \(\alpha\)-diversity measures.
# Plot alpha-diversity
plot_richness(ps, x="Day", measures = c("Shannon", "Simpson"), color =
"When") +
theme_bw()## Warning in estimate_richness(physeq, split = TRUE, measures = measures): The data you have provided does not have
## any singletons. This is highly suspicious. Results of richness
## estimates (for example) are probably unreliable, or wrong, if you have already
## trimmed low-abundance taxa from the data.
##
## We recommended that you find the un-trimmed data and retry.
Ordinate
Another type of diversity is that between units: how dissimilar or different are they? Ecologists will do what is called ordination in which they will assess distances or dissimilarities between individuals, then describe the variability in those assessments. Recall our last lesson in which we talked about non-metrical Multidimensional Scaling (nMDS). This is just one applicaiton of nMDS.
Let’s see how these fall when we ordinate using the Bray-Curtis dissimilarity index.
# Ordinate with Bray-Curtis
ord.nmds.bray <- ordinate(ps, method="NMDS", distance="bray")## Square root transformation
## Wisconsin double standardization
## Run 0 stress 8.398672e-05
## Run 1 stress 9.583921e-05
## ... Procrustes: rmse 0.0001495836 max resid 0.0002898768
## ... Similar to previous best
## Run 2 stress 8.952235e-05
## ... Procrustes: rmse 0.0001421689 max resid 0.0002888046
## ... Similar to previous best
## Run 3 stress 9.642257e-05
## ... Procrustes: rmse 3.184668e-05 max resid 5.311138e-05
## ... Similar to previous best
## Run 4 stress 8.690212e-05
## ... Procrustes: rmse 0.0001273792 max resid 0.0001784601
## ... Similar to previous best
## Run 5 stress 9.981066e-05
## ... Procrustes: rmse 0.0001631741 max resid 0.0003138411
## ... Similar to previous best
## Run 6 stress 9.922992e-05
## ... Procrustes: rmse 3.585768e-05 max resid 5.877498e-05
## ... Similar to previous best
## Run 7 stress 9.521692e-05
## ... Procrustes: rmse 8.151916e-05 max resid 0.0002688704
## ... Similar to previous best
## Run 8 stress 9.72407e-05
## ... Procrustes: rmse 3.610421e-05 max resid 6.086787e-05
## ... Similar to previous best
## Run 9 stress 8.12164e-05
## ... New best solution
## ... Procrustes: rmse 0.0001087098 max resid 0.0002434832
## ... Similar to previous best
## Run 10 stress 9.555921e-05
## ... Procrustes: rmse 0.000105762 max resid 0.0001927576
## ... Similar to previous best
## Run 11 stress 9.670001e-05
## ... Procrustes: rmse 0.0001354074 max resid 0.0003026794
## ... Similar to previous best
## Run 12 stress 9.744932e-05
## ... Procrustes: rmse 4.187815e-05 max resid 9.423736e-05
## ... Similar to previous best
## Run 13 stress 9.763069e-05
## ... Procrustes: rmse 0.0001418992 max resid 0.0002645107
## ... Similar to previous best
## Run 14 stress 9.525835e-05
## ... Procrustes: rmse 9.638446e-05 max resid 0.0002197075
## ... Similar to previous best
## Run 15 stress 9.84046e-05
## ... Procrustes: rmse 5.001935e-05 max resid 0.0001313918
## ... Similar to previous best
## Run 16 stress 8.344395e-05
## ... Procrustes: rmse 0.0001088384 max resid 0.0002210162
## ... Similar to previous best
## Run 17 stress 9.826413e-05
## ... Procrustes: rmse 0.0001360471 max resid 0.0002789851
## ... Similar to previous best
## Run 18 stress 9.928382e-05
## ... Procrustes: rmse 0.0001365506 max resid 0.0002790687
## ... Similar to previous best
## Run 19 stress 9.700253e-05
## ... Procrustes: rmse 7.633414e-05 max resid 0.0002058908
## ... Similar to previous best
## Run 20 stress 9.754811e-05
## ... Procrustes: rmse 0.000135635 max resid 0.0002740011
## ... Similar to previous best
## *** Solution reached## Warning in metaMDS(veganifyOTU(physeq), distance, ...): Stress is (nearly)
## zero - you may have insufficient dataplot_ordination(ps, ord.nmds.bray, color="When", title="Bray NMDS")
We see that ordination picks out a separation between the early and late samples.
Bar Plots
Another common practice in microbiome research is to determine the top N categories at some taxonomic level. One of my collaborators calls this the production of the “Greatest Hits.”
Let’s pick out the top 20 OTUs, then see how they fall in individuals, colored by Family, and grouped by early or late.
# Create bar plots for top 20 OTUs
top20 <- names(sort(taxa_sums(ps), decreasing = TRUE))[1:20]
ps.top20 <- transform_sample_counts(ps, function(OTU) OTU/sum(OTU))
ps.top20 <- prune_taxa(top20, ps.top20)
plot_bar(ps.top20, x="Day", fill="Family") + facet_wrap(~When, scales="free_x")## Warning: Removed 20 rows containing missing values (position_stack).
That wraps it up for today. Next week we will show more phyloseq and then get into functional predictions.