Comparing predicted UTRs
Philipp Ross
2018-09-24
Last updated: 2018-09-25
Code version: 9220caa
Comparing different UTR and TSS predictions
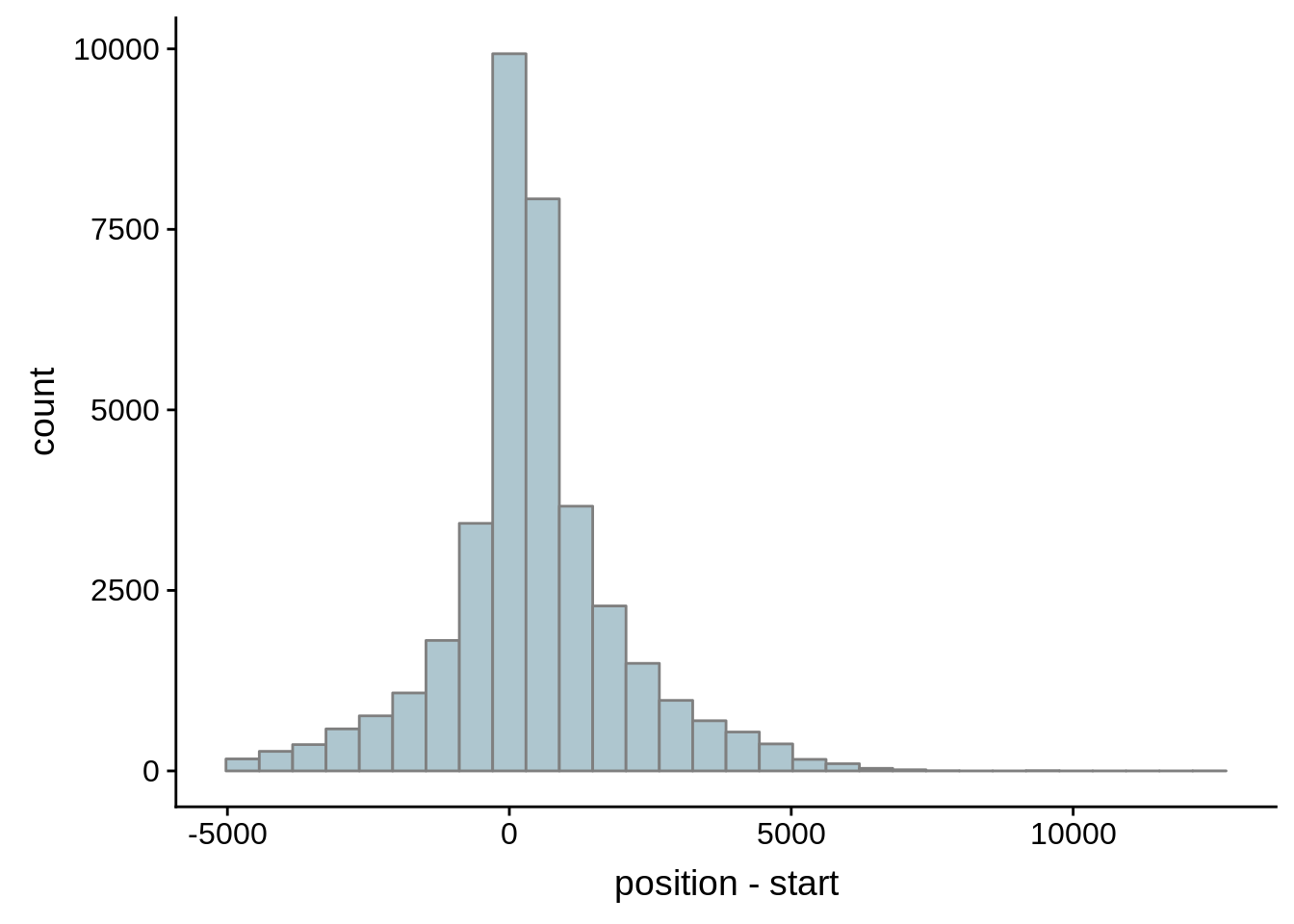
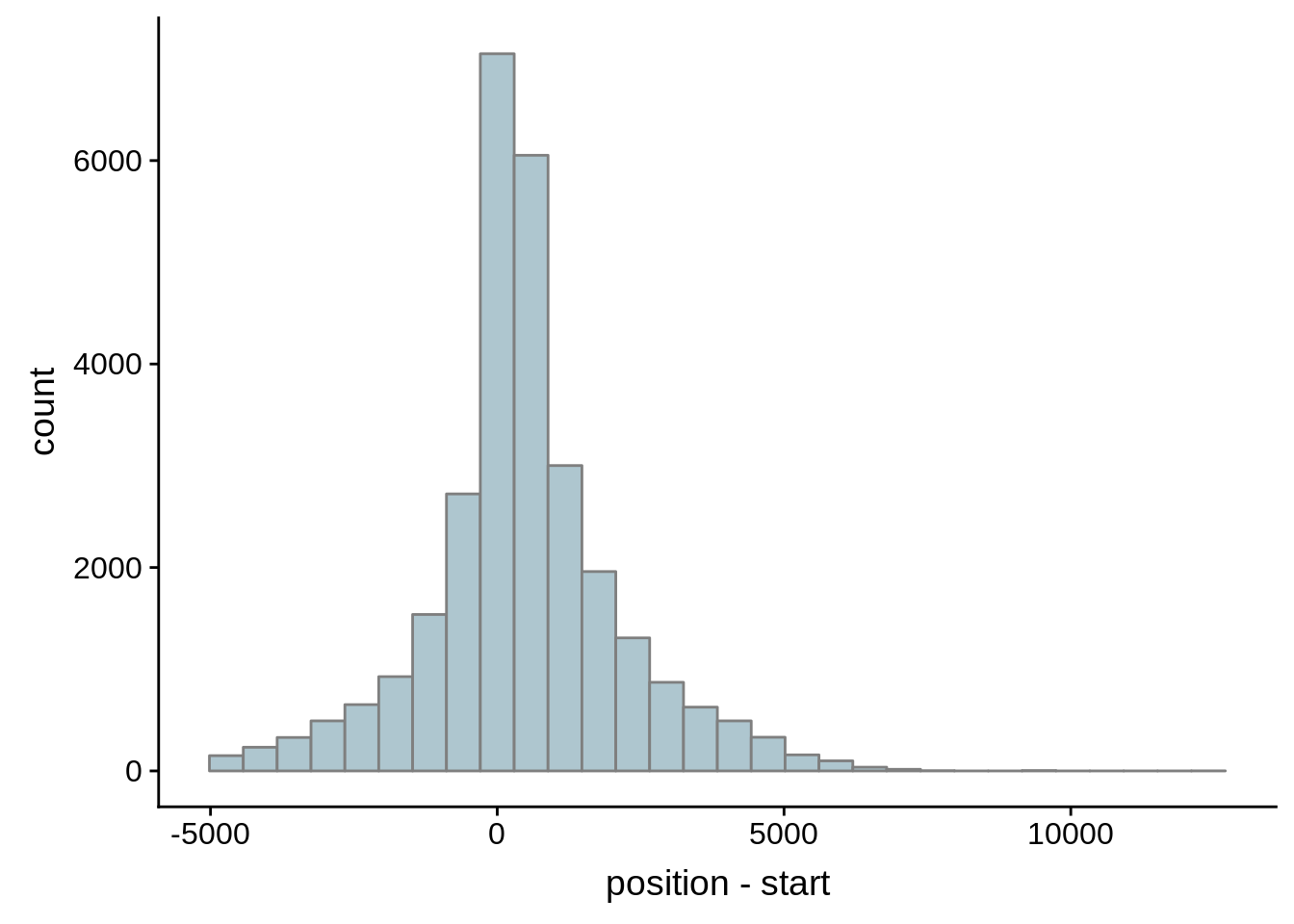
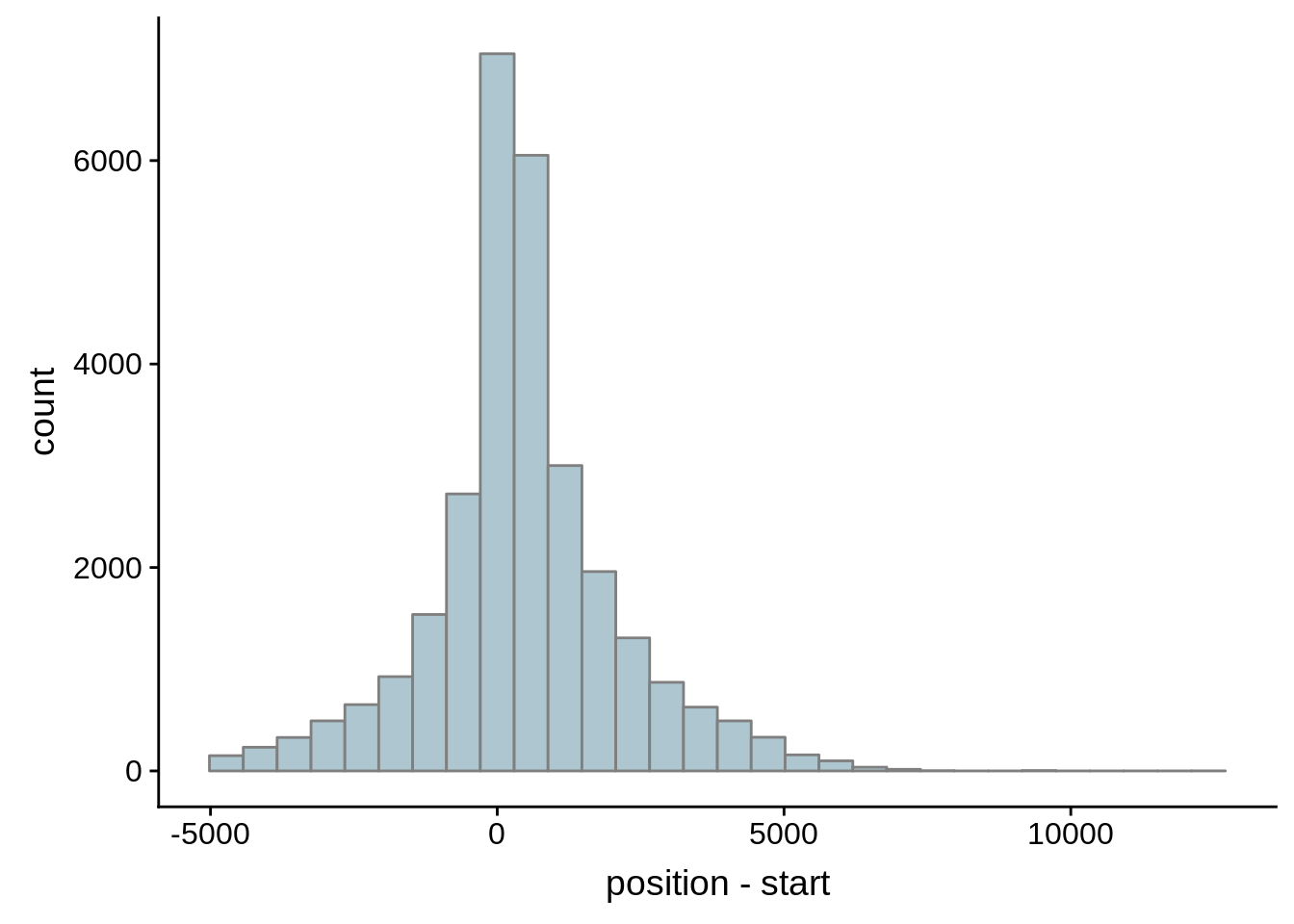
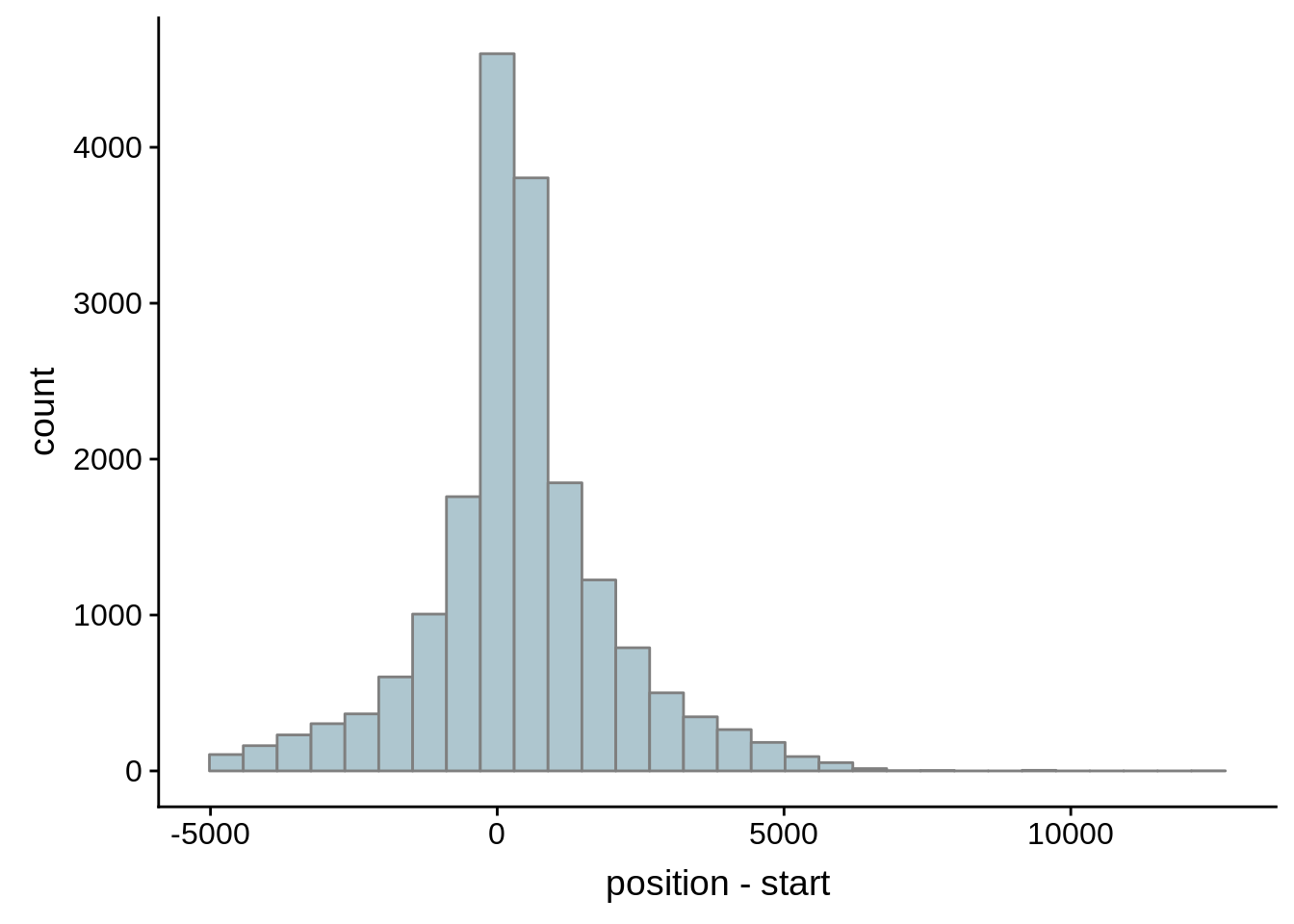
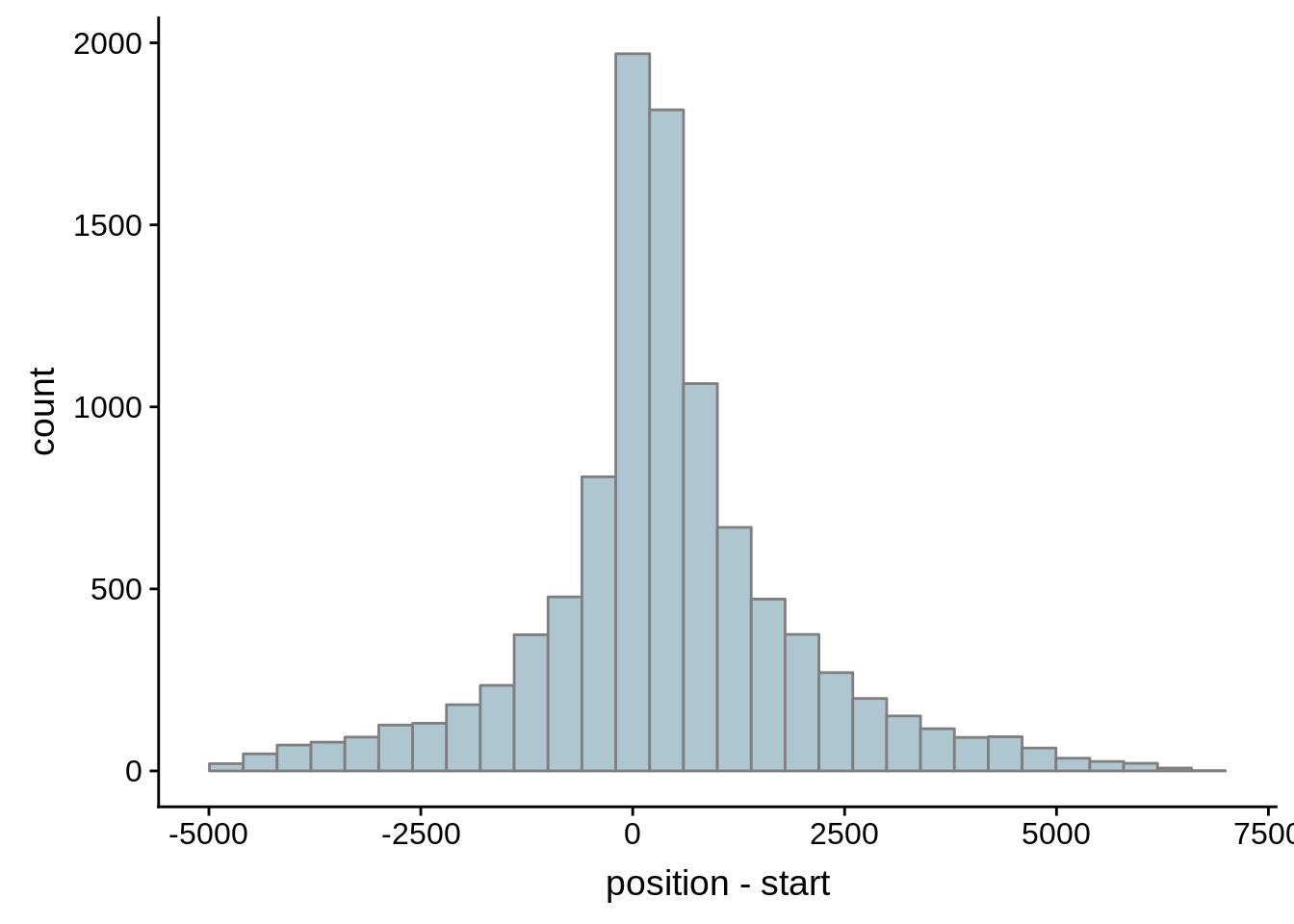
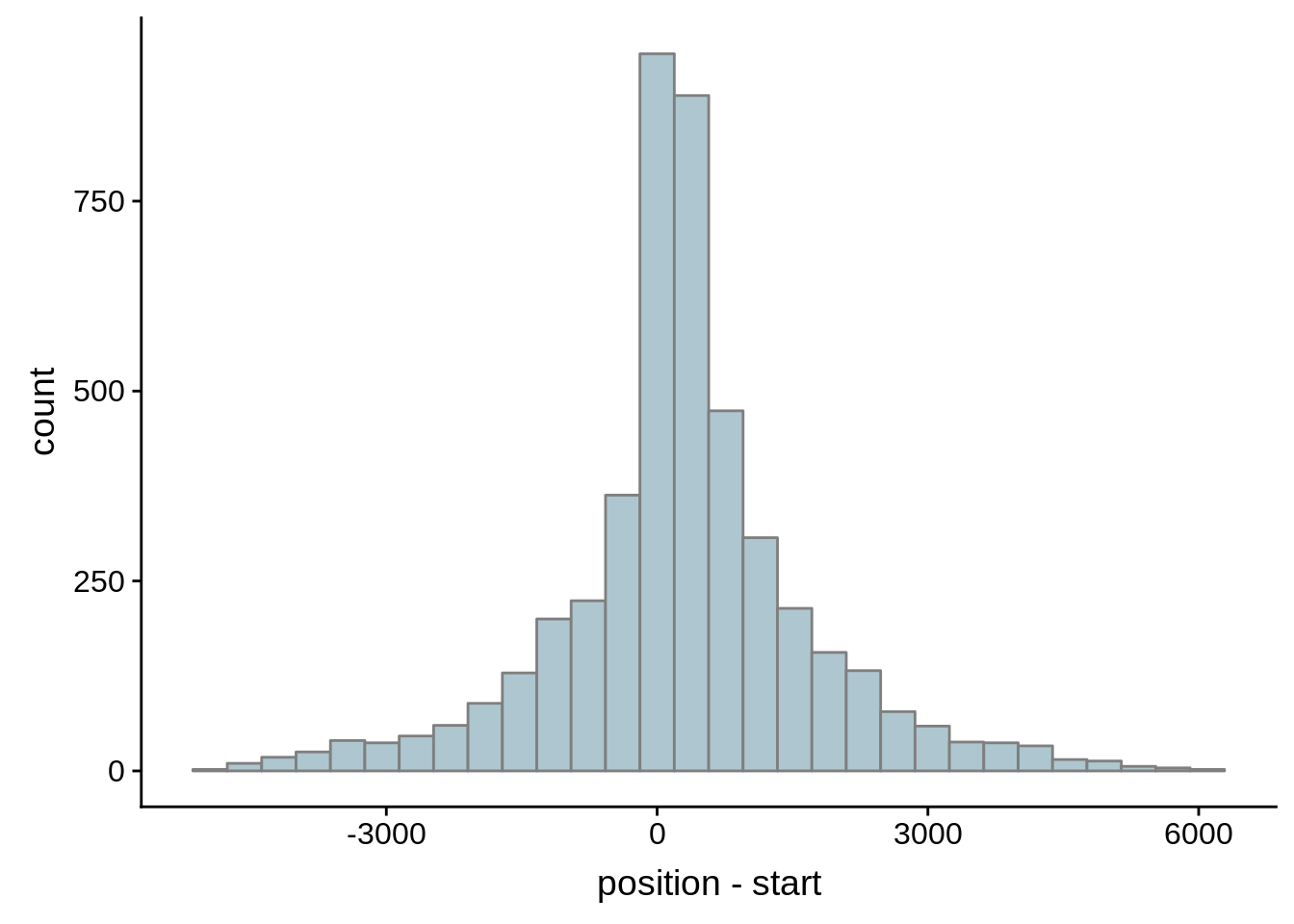
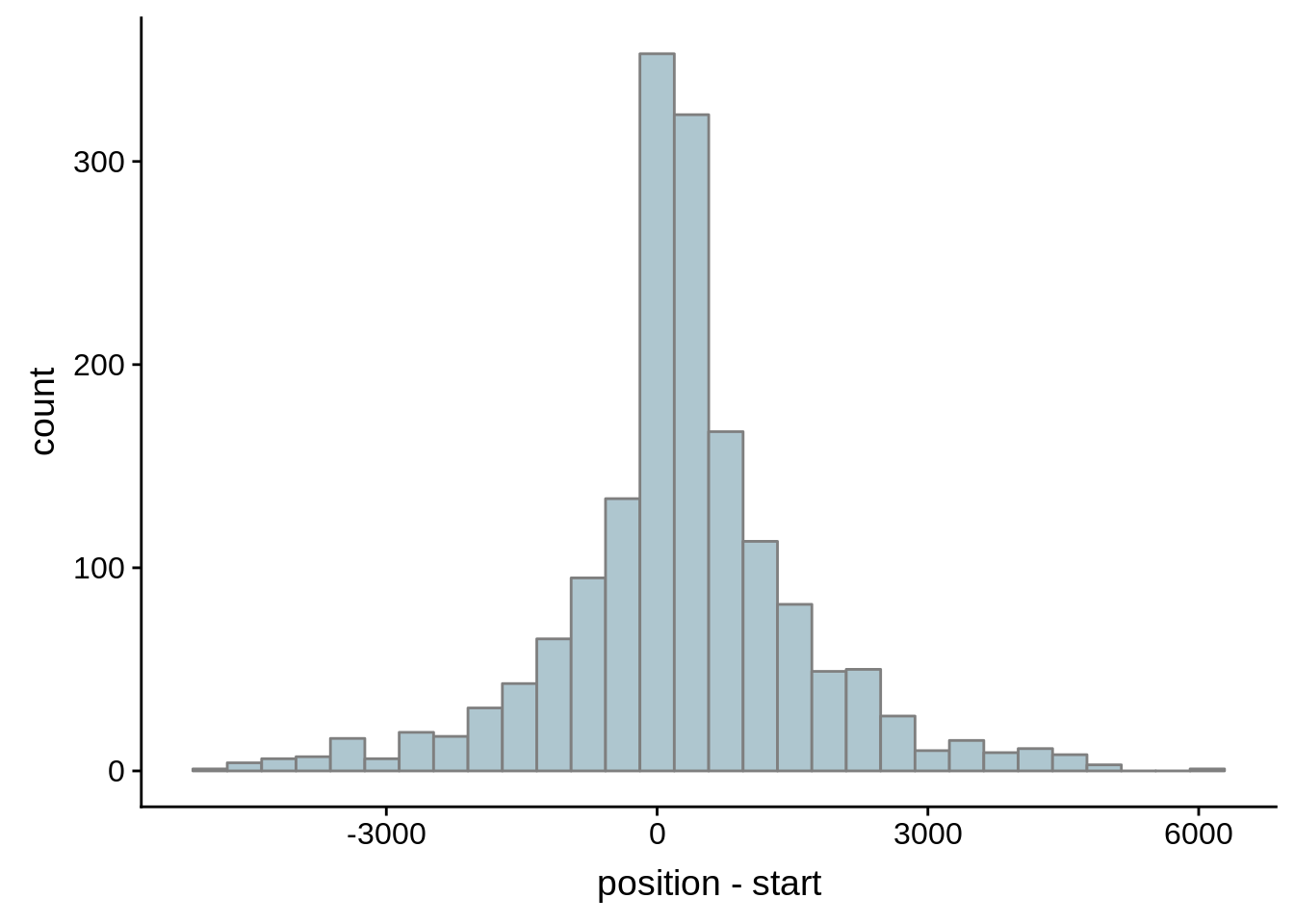
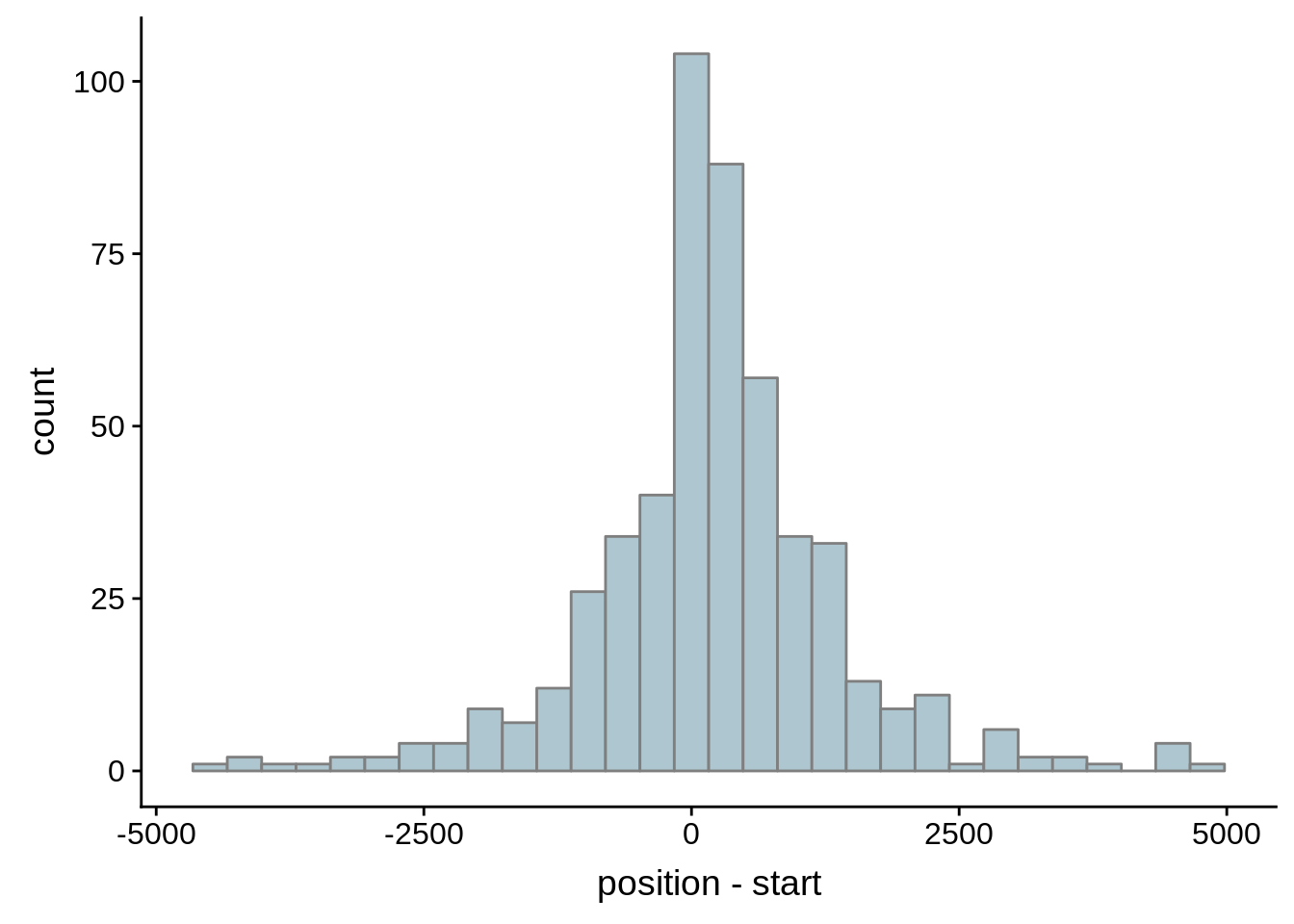
Since we were able to predict both UTRs and TSSs using our data, we wanted to know how our predictions compared to previously published predictions. Here, we compare the UTRs predicted in Caro et al. and Adjalley et al..
What we see is that although there are some large deviations, for the majority of 5UTR and TSS predictions, the results are not very different with a mean hovering around zero base pairs of difference between the start positions of our predicted 5’ UTRs and TSS and those that were previously published.
First, let’s import our 5UTR data:
library(org.Pf.plasmo.db)
aliases <- tibble::as_tibble(data.frame(org.Pf.plasmoALIAS2ORF))
utrs <- tibble::as_tibble(rtracklayer::import.gff("../output/final_utrs/final_5utrs_3d7.gff"))
utrs$Parent <- unlist(utrs$Parent)Comparing to Caro et al.
# Comparing our UTR estimates to the Derisi predictions
derisi1 <- tibble::as_tibble(rtracklayer::import.bed("../data/compare_utrs/GSM1410291_UTRs_1.bed"))
derisi2 <- tibble::as_tibble(rtracklayer::import.bed("../data/compare_utrs/GSM1410292_UTRs_2.bed"))
derisi3 <- tibble::as_tibble(rtracklayer::import.bed("../data/compare_utrs/GSM1410293_UTRs_3.bed"))
derisi4 <- tibble::as_tibble(rtracklayer::import.bed("../data/compare_utrs/GSM1410294_UTRs_4.bed"))
derisi5 <- tibble::as_tibble(rtracklayer::import.bed("../data/compare_utrs/GSM1410295_UTRs_5.bed"))
fix_derisi_utrs <- function(set) {
set$name <- stringi::stri_replace_last(set$name,replacement=" ",regex="_")
set <- set %>% tidyr::separate(name,into = c("gene_id","type"), sep =" ")
set$gene_id <- toupper(set$gene_id)
out <- dplyr::inner_join(set, aliases, by=c("gene_id"="alias_symbol"))
return(out)
}
derisi1 <- fix_derisi_utrs(derisi1)
derisi2 <- fix_derisi_utrs(derisi2)
derisi3 <- fix_derisi_utrs(derisi3)
derisi4 <- fix_derisi_utrs(derisi4)
derisi5 <- fix_derisi_utrs(derisi5)tmp1 <- dplyr::select(derisi1, gene_id.y, width)
tmp2 <- dplyr::select(utrs, Parent, width)
compare_derisi <- dplyr::inner_join(tmp1,tmp2,by=c("gene_id.y"="Parent"))
rm(tmp1,tmp2)
ggplot(compare_derisi,aes(width.x,width.y)) + geom_point() + geom_smooth(method="lm")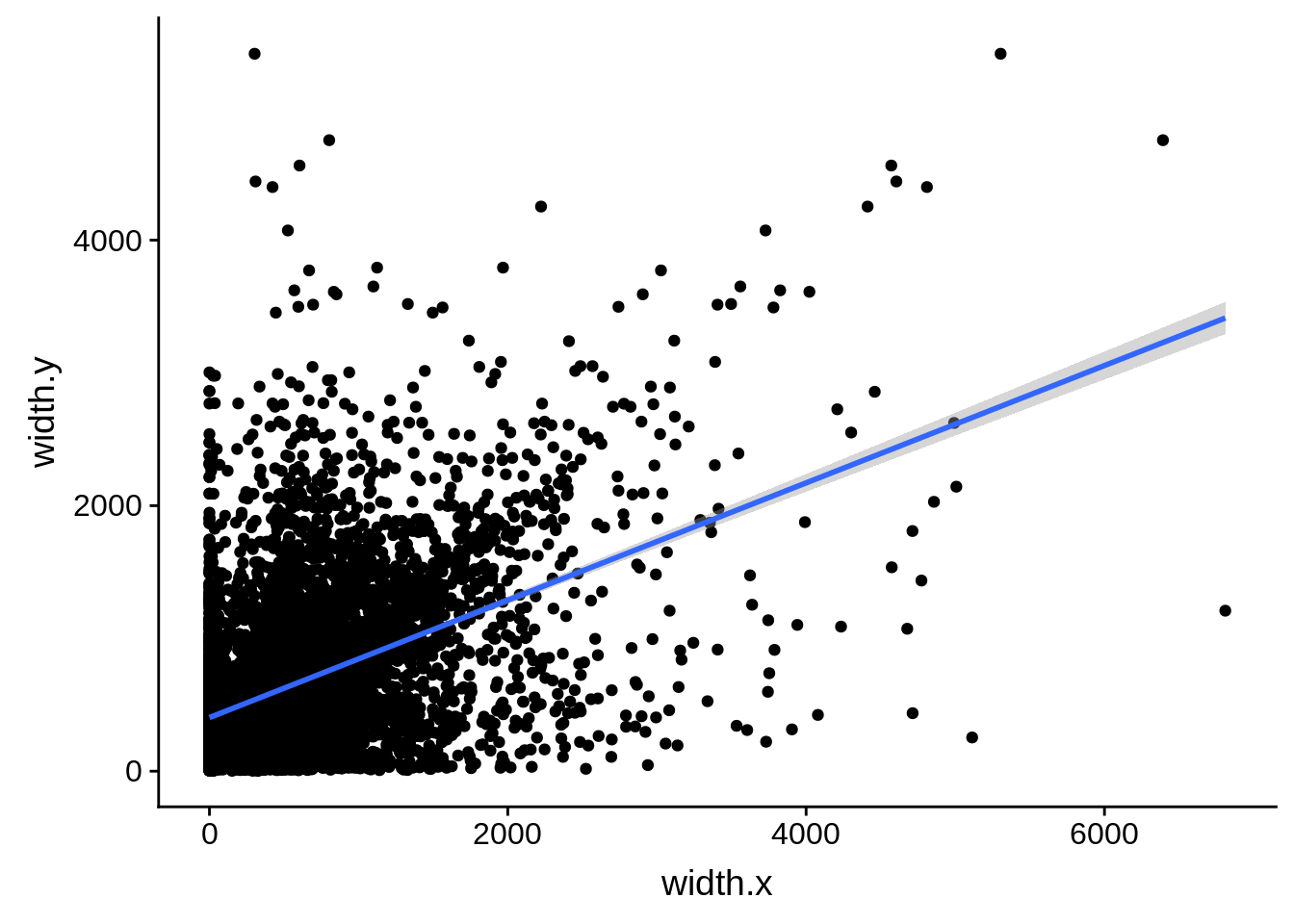
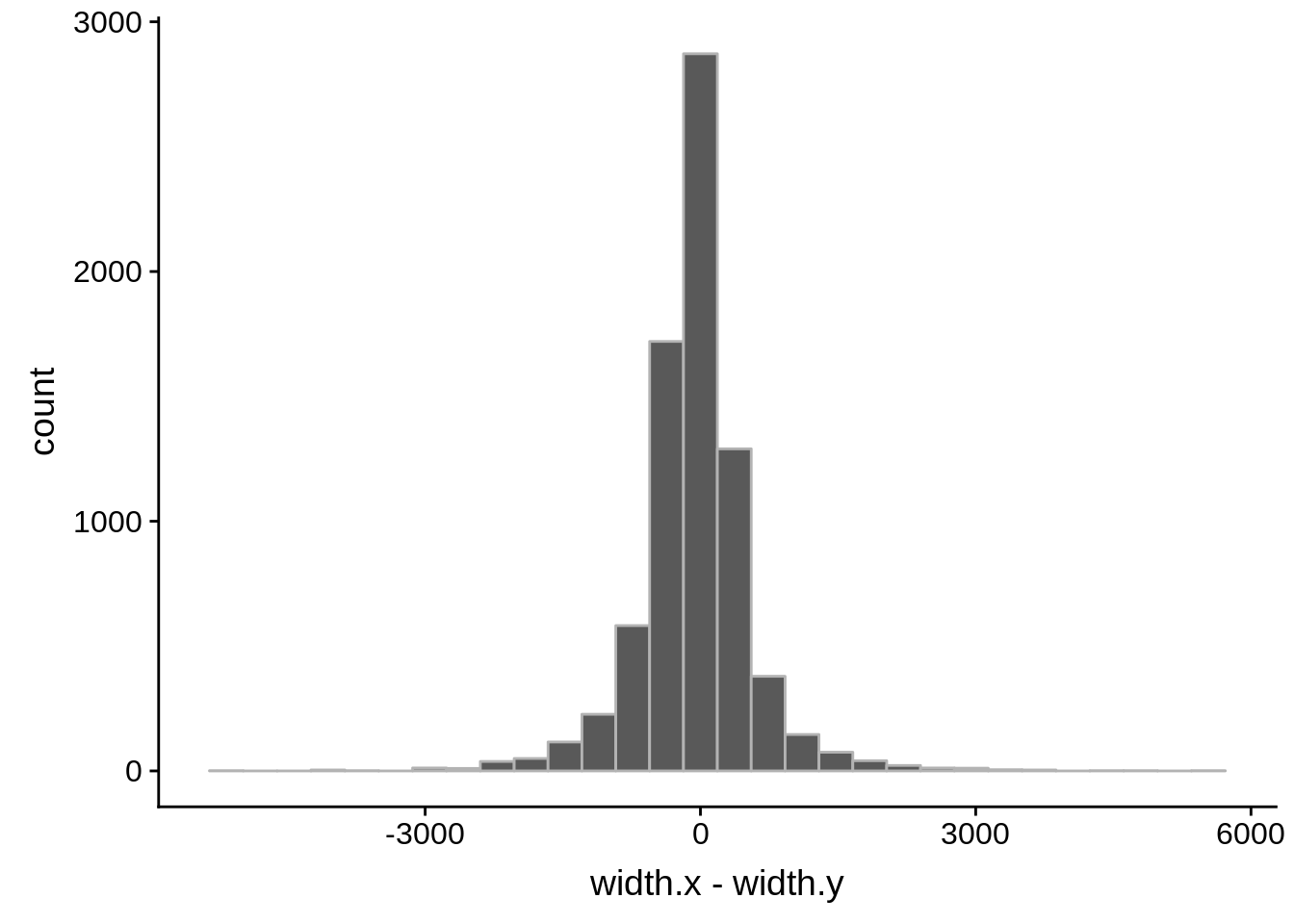
tmp1 <- dplyr::select(derisi2, gene_id.y, width)
tmp2 <- dplyr::select(utrs, Parent, width)
compare_derisi <- dplyr::inner_join(tmp1,tmp2,by=c("gene_id.y"="Parent"))
rm(tmp1,tmp2)
ggplot(compare_derisi,aes(width.x,width.y)) + geom_point() + geom_smooth(method="lm")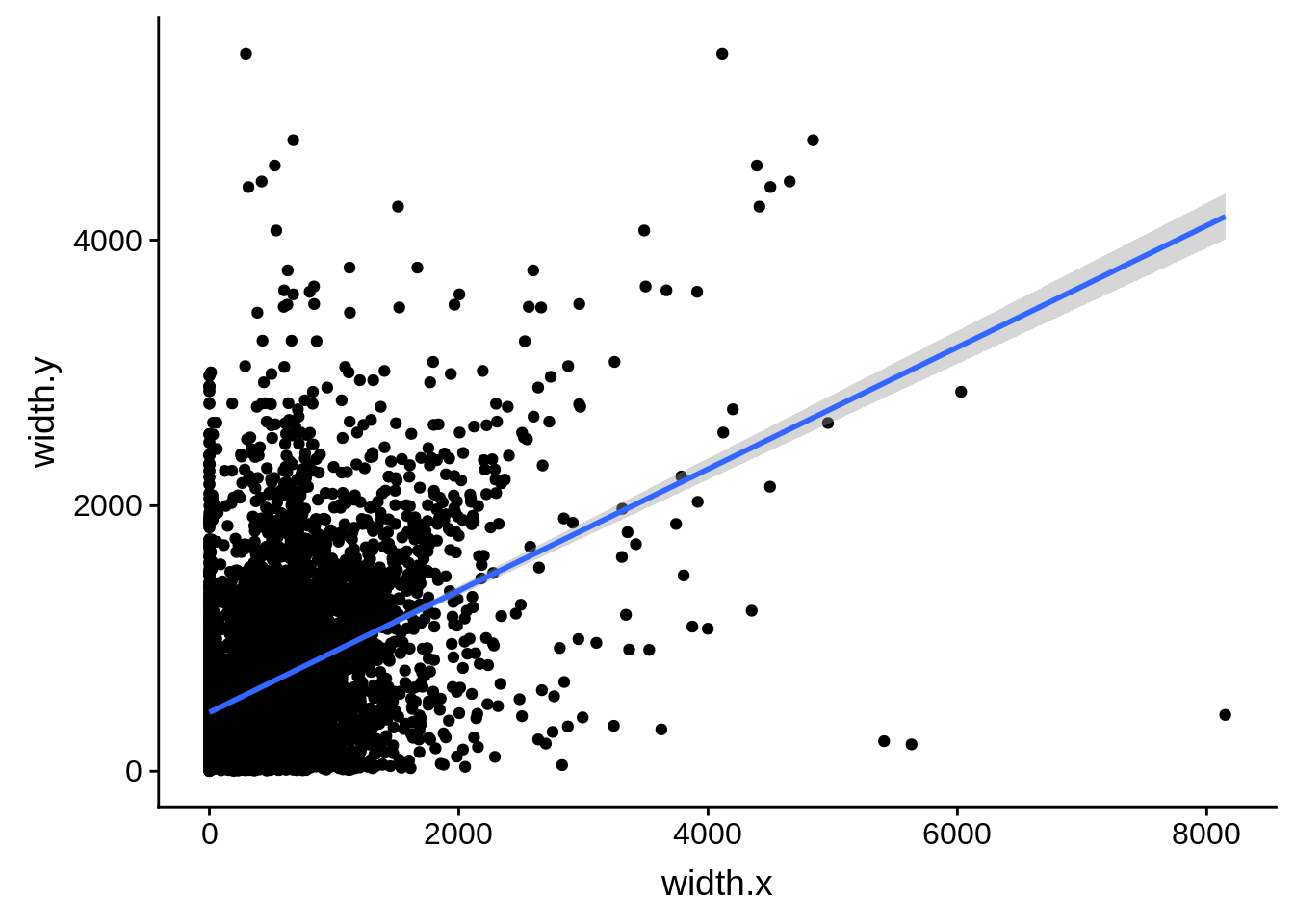
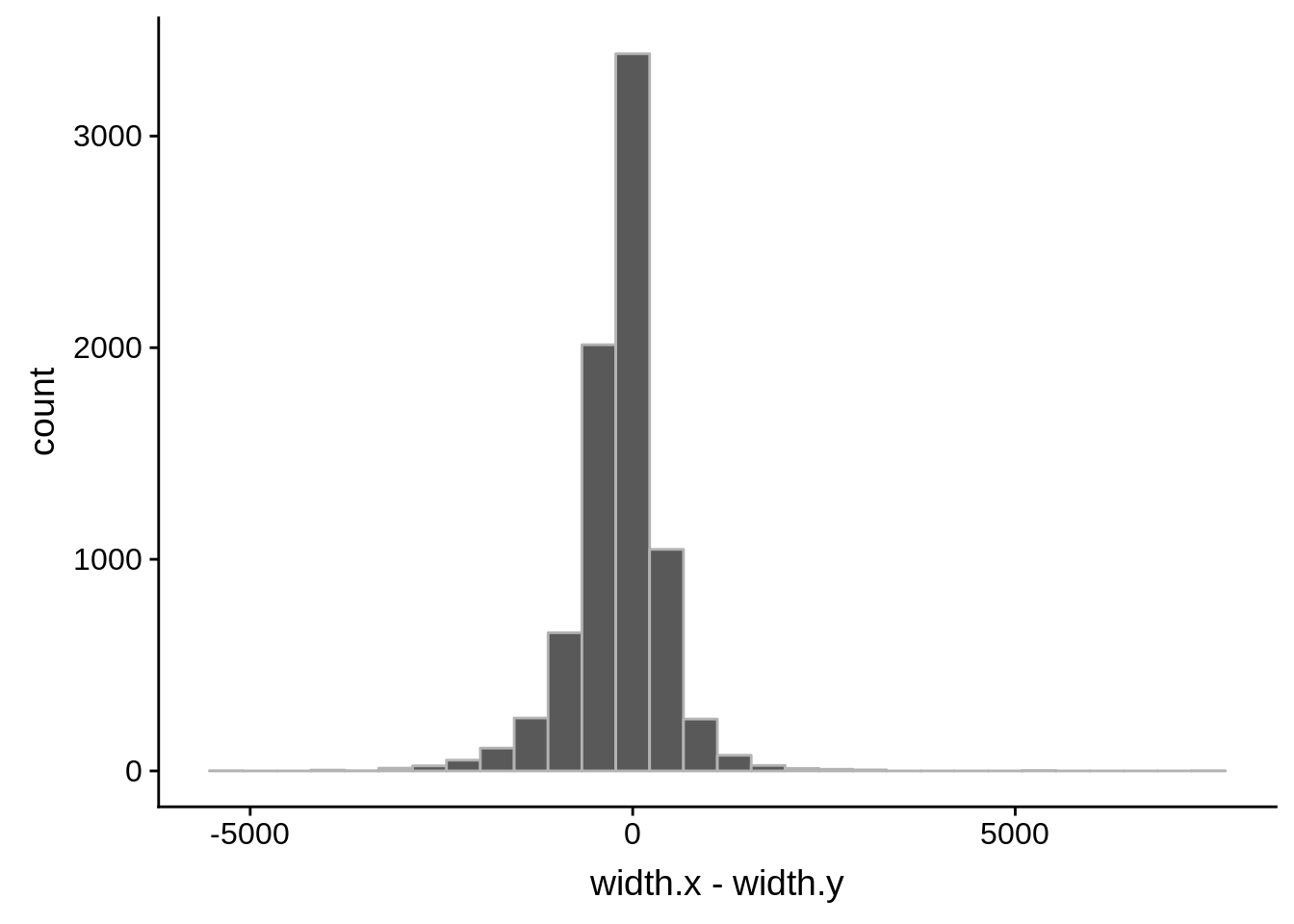
tmp1 <- dplyr::select(derisi3, gene_id.y, width)
tmp2 <- dplyr::select(utrs, Parent, width)
compare_derisi <- dplyr::inner_join(tmp1,tmp2,by=c("gene_id.y"="Parent"))
rm(tmp1,tmp2)
ggplot(compare_derisi,aes(width.x,width.y)) + geom_point() + geom_smooth(method="lm")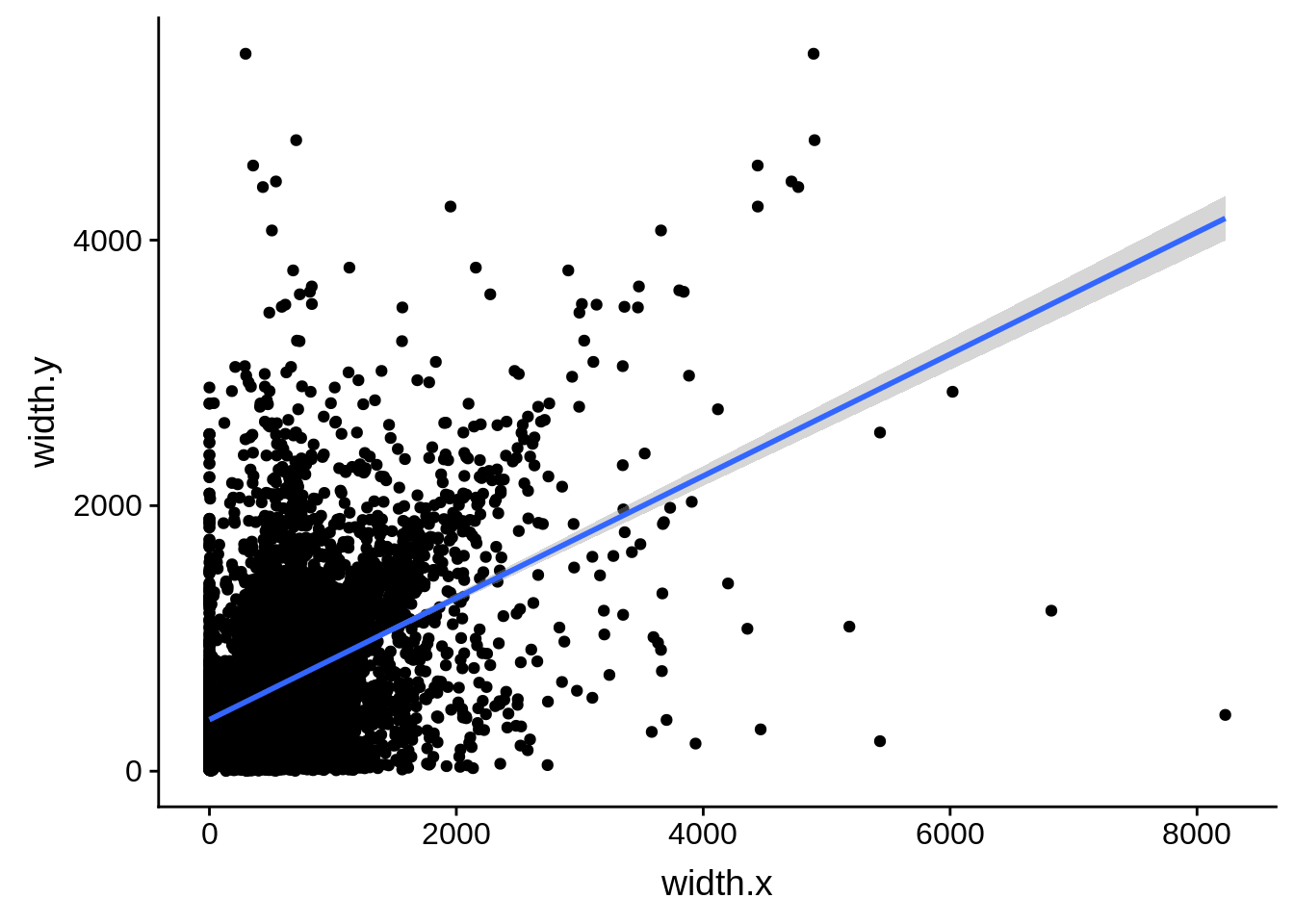
tmp1 <- dplyr::select(derisi3, gene_id.y, width)
tmp2 <- dplyr::select(utrs, Parent, width)
compare_derisi <- dplyr::inner_join(tmp1,tmp2,by=c("gene_id.y"="Parent"))
rm(tmp1,tmp2)
ggplot(compare_derisi,aes(width.x,width.y)) + geom_point() + geom_smooth(method="lm")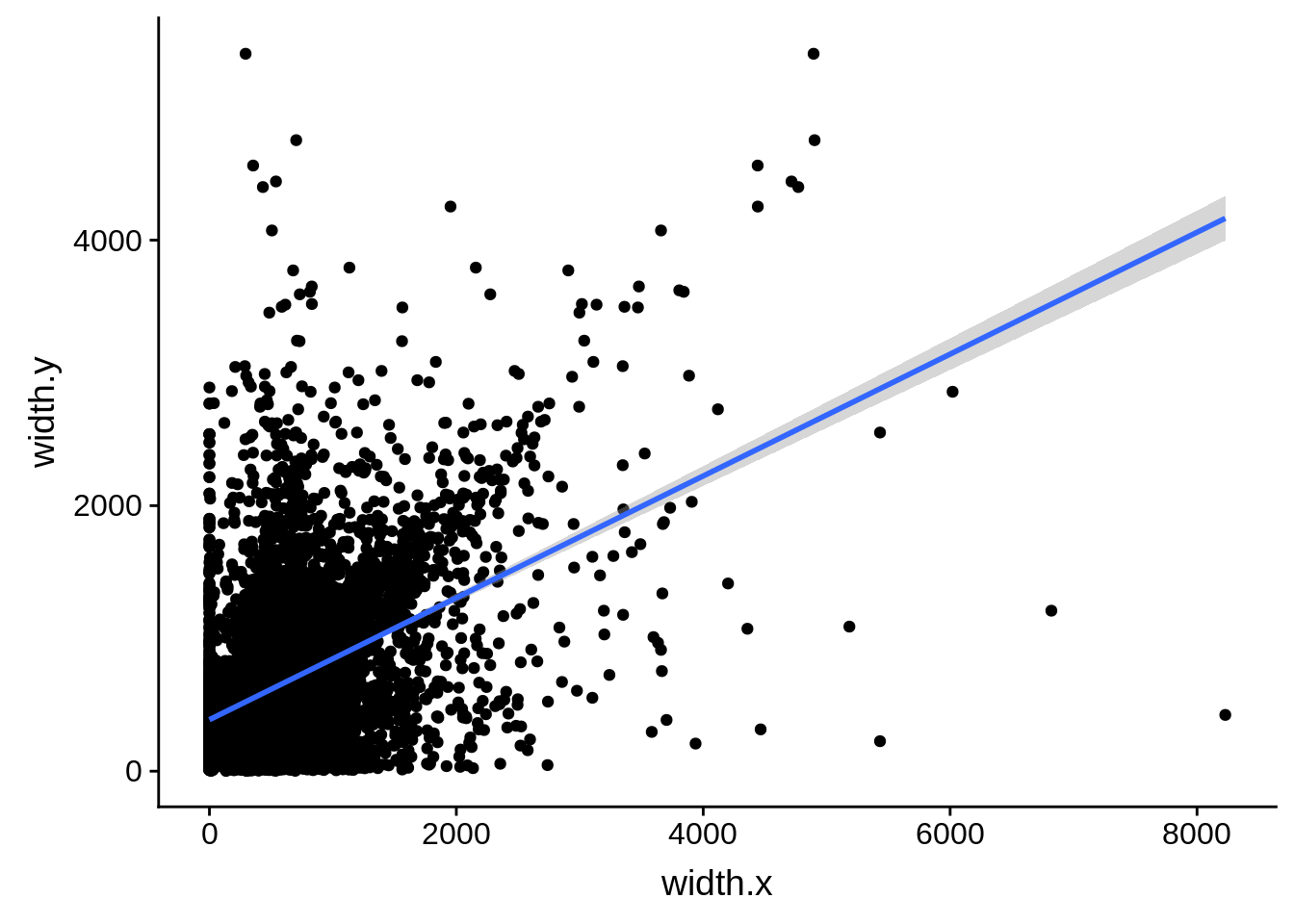
tmp1 <- dplyr::select(derisi4, gene_id.y, width)
tmp2 <- dplyr::select(utrs, Parent, width)
compare_derisi <- dplyr::inner_join(tmp1,tmp2,by=c("gene_id.y"="Parent"))
rm(tmp1,tmp2)
ggplot(compare_derisi,aes(width.x,width.y)) + geom_point() + geom_smooth(method="lm")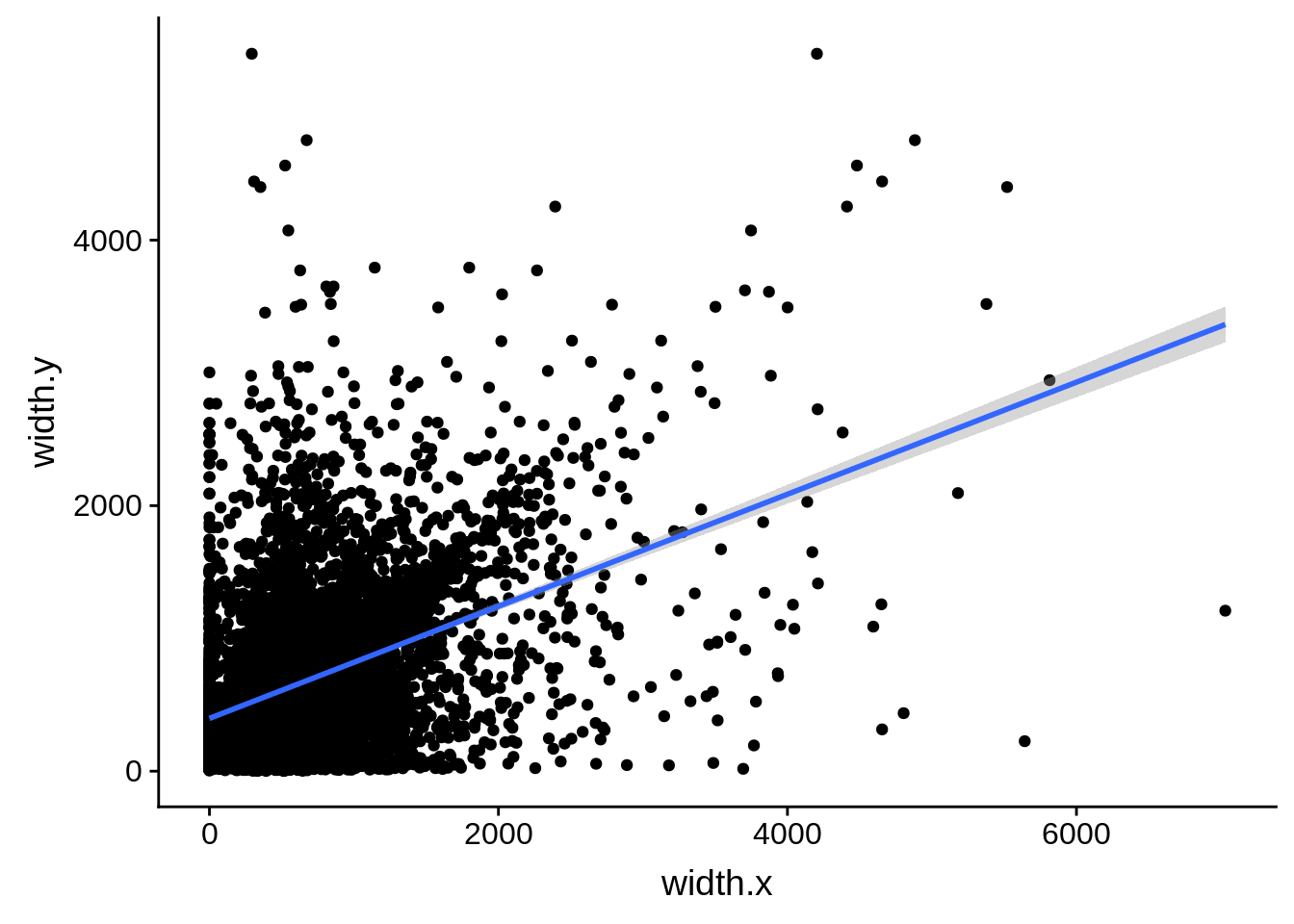
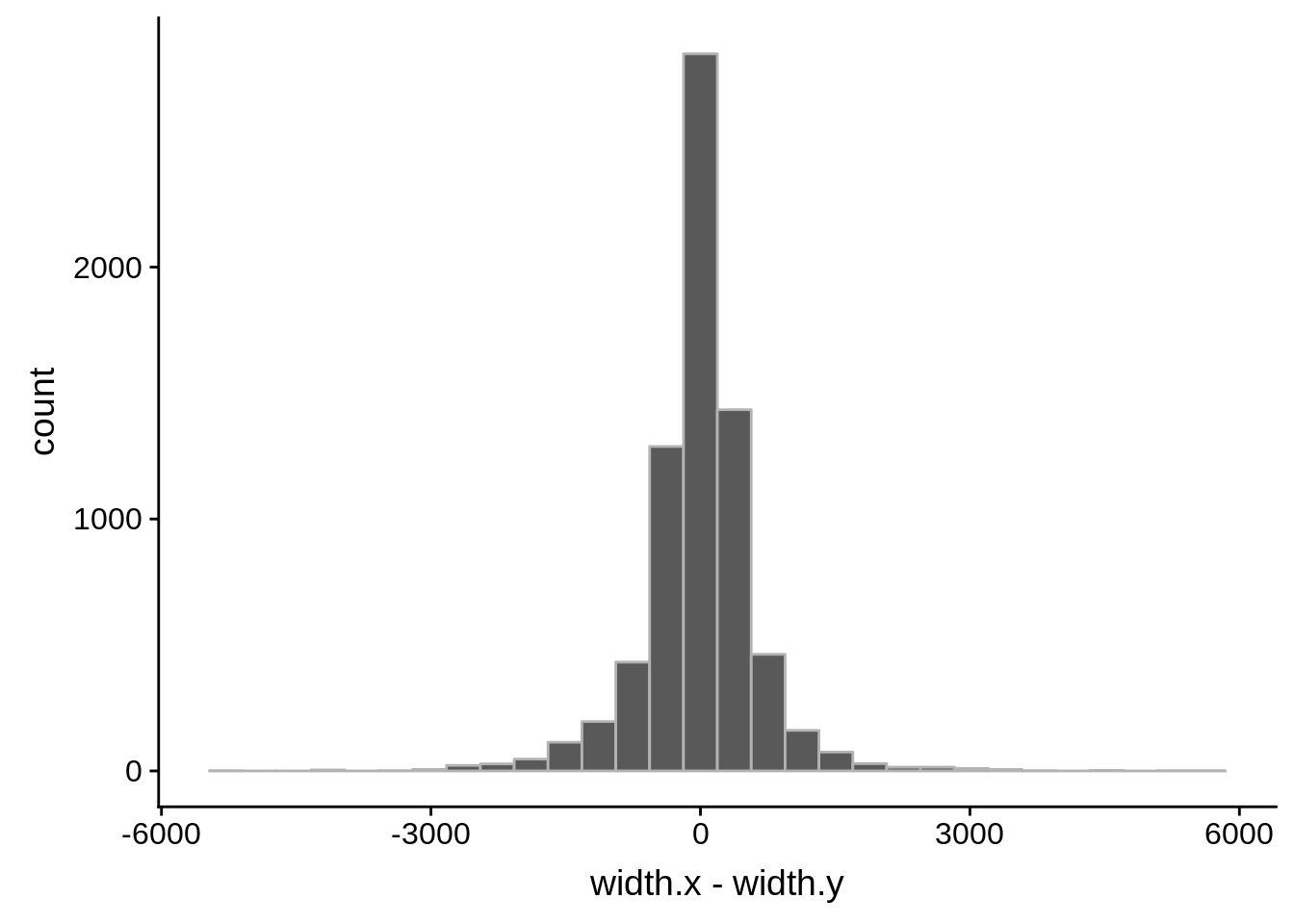
tmp1 <- dplyr::select(derisi5, gene_id.y, width)
tmp2 <- dplyr::select(utrs, Parent, width)
compare_derisi <- dplyr::inner_join(tmp1,tmp2,by=c("gene_id.y"="Parent"))
rm(tmp1,tmp2)
ggplot(compare_derisi,aes(width.x,width.y)) + geom_point() + geom_smooth(method="lm")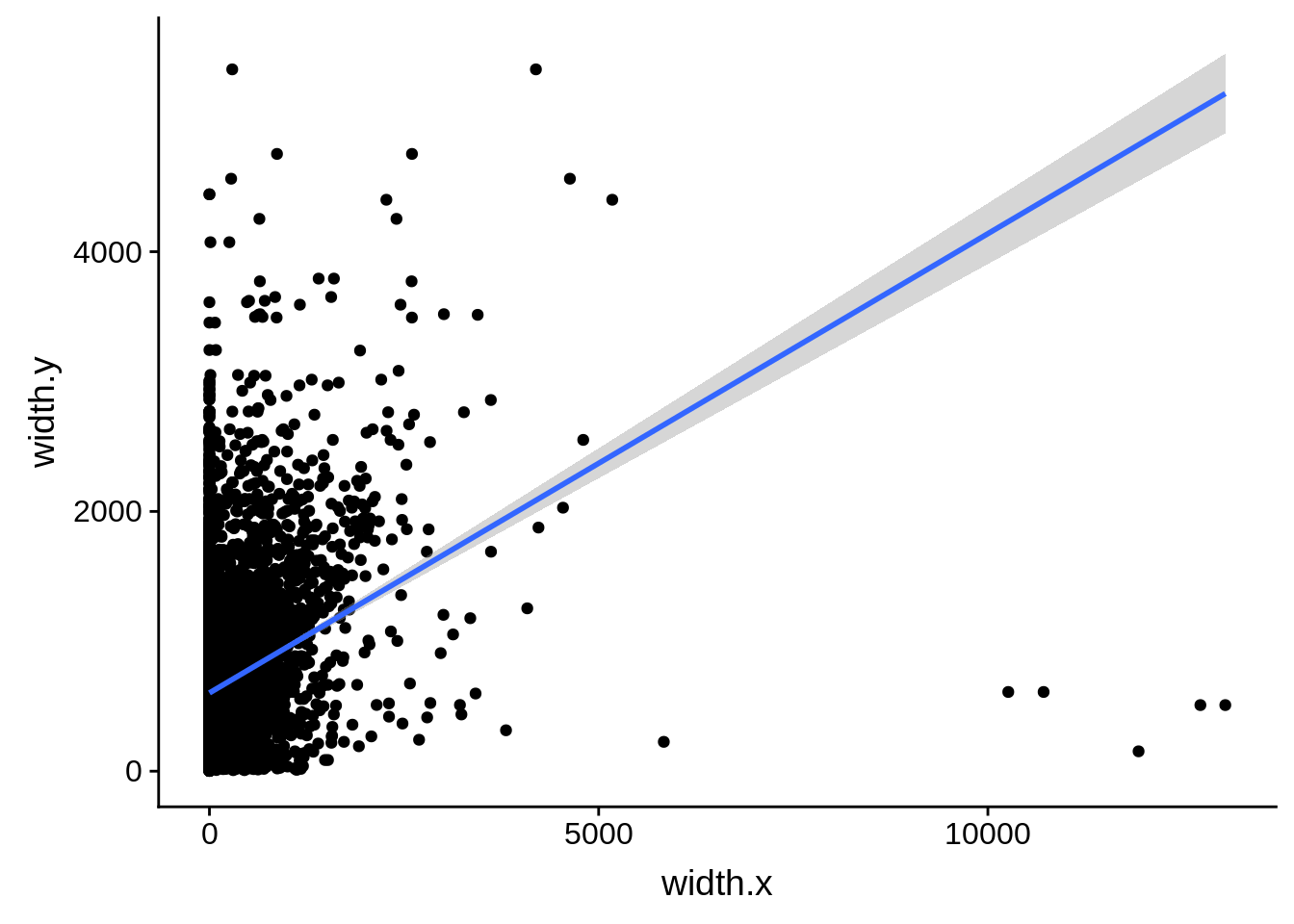
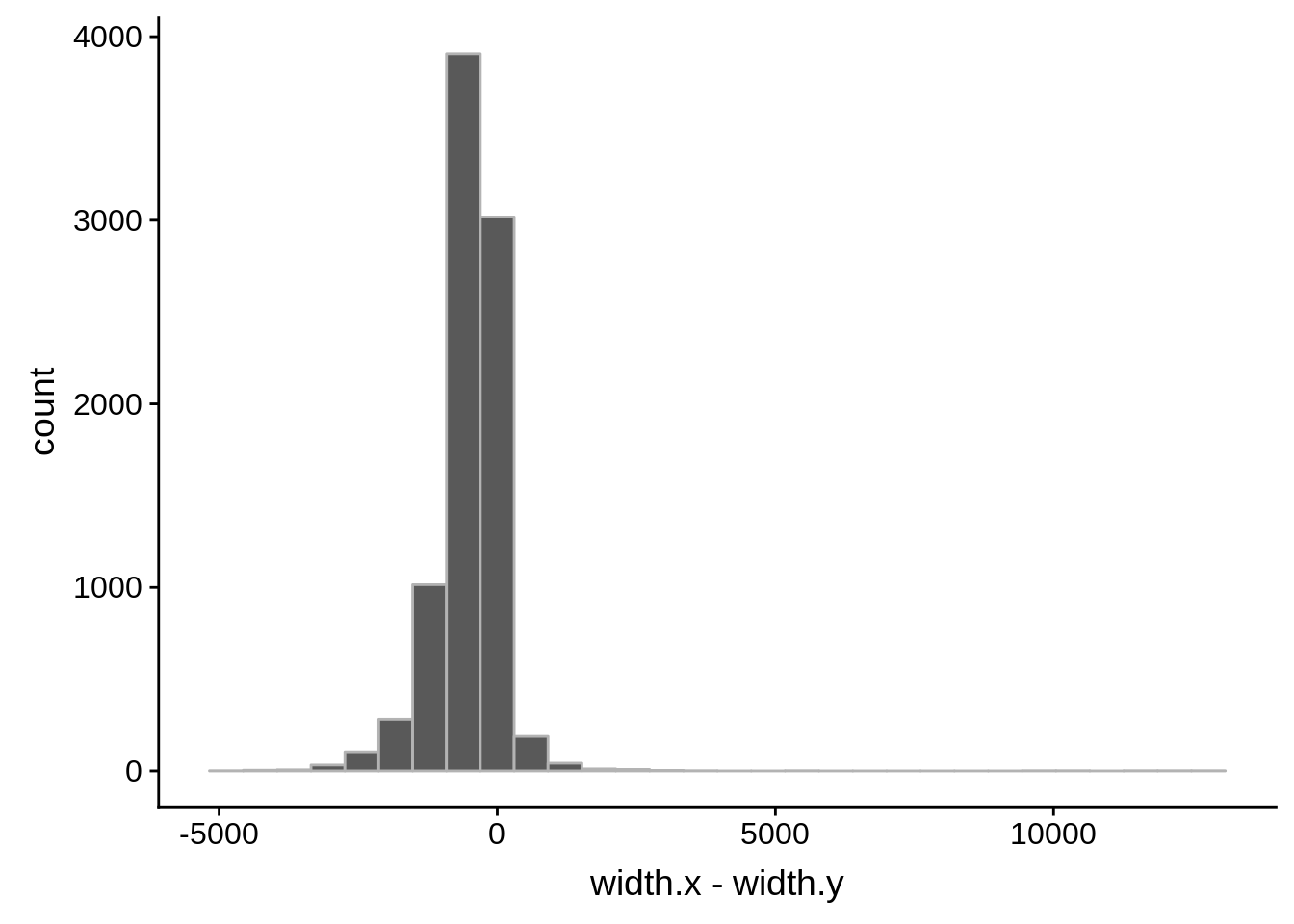
Comparing to Adjalley et al.
# Comparing our UTR estimates to the Adjalley predictions
adjalley <- tibble::as_tibble(rtracklayer::import.gff("../data/compare_utrs/sorted_Adjalley_Chabbert_TSSs.gff"))
for (i in 1:8) {
adjalley_filtered <- adjalley %>%
dplyr::mutate(position=(start+end)/2) %>%
dplyr::filter(FilterSize>i)
tmp1 <- dplyr::select(adjalley_filtered, AssignedFeat, position)
tmp2 <- dplyr::select(utrs, Parent, start)
compare_adjalley <- dplyr::inner_join(tmp1,tmp2,by=c("AssignedFeat"="Parent"))
g <- ggplot(compare_adjalley,aes(position-start)) + geom_histogram(fill="#aec6cf",color="grey50")
print(g)
}
Session Information
R version 3.5.0 (2018-04-23)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Gentoo/Linux
Matrix products: default
BLAS: /usr/local/lib64/R/lib/libRblas.so
LAPACK: /usr/local/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] bindrcpp_0.2.2
[2] BSgenome.Pfalciparum.PlasmoDB.v24_1.0
[3] BSgenome_1.48.0
[4] rtracklayer_1.40.6
[5] Biostrings_2.48.0
[6] XVector_0.20.0
[7] GenomicRanges_1.32.6
[8] GenomeInfoDb_1.16.0
[9] org.Pf.plasmo.db_3.6.0
[10] AnnotationDbi_1.42.1
[11] IRanges_2.14.10
[12] S4Vectors_0.18.3
[13] Biobase_2.40.0
[14] BiocGenerics_0.26.0
[15] scales_1.0.0
[16] cowplot_0.9.3
[17] magrittr_1.5
[18] forcats_0.3.0
[19] stringr_1.3.1
[20] dplyr_0.7.6
[21] purrr_0.2.5
[22] readr_1.1.1
[23] tidyr_0.8.1
[24] tibble_1.4.2
[25] ggplot2_3.0.0
[26] tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] nlme_3.1-137 bitops_1.0-6
[3] matrixStats_0.54.0 lubridate_1.7.4
[5] bit64_0.9-7 httr_1.3.1
[7] rprojroot_1.3-2 tools_3.5.0
[9] backports_1.1.2 R6_2.2.2
[11] DBI_1.0.0 lazyeval_0.2.1
[13] colorspace_1.3-2 withr_2.1.2
[15] tidyselect_0.2.4 bit_1.1-14
[17] compiler_3.5.0 git2r_0.23.0
[19] cli_1.0.0 rvest_0.3.2
[21] xml2_1.2.0 DelayedArray_0.6.5
[23] labeling_0.3 digest_0.6.15
[25] Rsamtools_1.32.3 rmarkdown_1.10
[27] R.utils_2.6.0 pkgconfig_2.0.2
[29] htmltools_0.3.6 rlang_0.2.2
[31] readxl_1.1.0 rstudioapi_0.7
[33] RSQLite_2.1.1 bindr_0.1.1
[35] jsonlite_1.5 BiocParallel_1.14.2
[37] R.oo_1.22.0 RCurl_1.95-4.11
[39] GenomeInfoDbData_1.1.0 Matrix_1.2-14
[41] Rcpp_0.12.18 munsell_0.5.0
[43] R.methodsS3_1.7.1 stringi_1.2.4
[45] yaml_2.2.0 SummarizedExperiment_1.10.1
[47] zlibbioc_1.26.0 plyr_1.8.4
[49] grid_3.5.0 blob_1.1.1
[51] crayon_1.3.4 lattice_0.20-35
[53] haven_1.1.2 hms_0.4.2
[55] knitr_1.20 pillar_1.3.0
[57] XML_3.98-1.16 glue_1.3.0
[59] evaluate_0.11 modelr_0.1.2
[61] cellranger_1.1.0 gtable_0.2.0
[63] assertthat_0.2.0 broom_0.5.0
[65] GenomicAlignments_1.16.0 memoise_1.1.0
[67] workflowr_1.1.1 This R Markdown site was created with workflowr