Finalizing the classifer results
Joyce Hsiao
Last updated: 2018-07-12
Code version: fb90641
Gene lists
genes_list <- readRDS(file = "../data/results/results_topgenes.rds")
all_genes <- genes_list[length(genes_list)][[1]]
library(biomaRt)
ensembl <- useMart(biomart = "ensembl", dataset = "hsapiens_gene_ensembl")
symbols <- getBM(attributes = c("hgnc_symbol",'ensembl_gene_id'),
filters = c('ensembl_gene_id'),
values = all_genes,
mart = ensembl)
genes_list_symbols <- lapply(1:length(genes_list), function(i) {
ll <- genes_list[i][[1]]
#symbols[match(ll,symbols$ensembl_gene_id),]
symbs <- symbols[which(symbols$ensembl_gene_id %in% ll),]
non_symbs <- ll[which(!(ll %in% symbols$ensembl_gene_id))]
df_non_symbs <- data.frame(hgnc_symbol=NA,
ensembl_gene_id=non_symbs)
out <- rbind(symbs, df_non_symbs)
out <- out[match(ll,out$ensembl_gene_id),]
return(out)
})
names(genes_list_symbols) <- names(genes_list)
saveRDS(genes_list_symbols,
"../output/method-train-classifiers-genes.Rmd/genes_list_symbols.rds")genes_list <- readRDS(file = "../data/results/results_topgenes.rds")
genes_list_symbols <- readRDS("../output/method-train-classifiers-genes.Rmd/genes_list_symbols.rds")
seurat.genes <- readLines(
con = "../data/cellcycle-genes-previous-studies/seurat_cellcycle/regev_lab_cell_cycle_genes.txt")
seurat.genes <- list(s.genes=seurat.genes[1:43],
g2m.genes=seurat.genes[44:97])
tmp <- sapply(genes_list_symbols, function(x) sum(x$hgnc_symbol %in% unlist(seurat.genes)))
# par(mfrow=c(1,2))
# plot(x=sapply(genes_list, length),
# tmp,
# xlab="Number of cyclical genes",
# ylab="Number of Seurate genes (total 97)",
# main = "Seurat genes in our data")
# plot(x=sapply(genes_list, length),
# tmp/as.numeric(names(genes_list)),
# xlab="Number of cyclical genes",
# ylab="Proportion of Seurate genes",
# main = "Seurat genes in our data")
par(mfrow=c(1,2))
plot(x=sapply(genes_list, length)[1:27],
tmp[1:27],
xlab="Number of cyclical genes",
ylab="Number of Seurate genes (total 97)",
main = "Seurat genes in top 260")
plot(x=sapply(genes_list, length)[1:27],
(tmp/as.numeric(names(genes_list)))[1:27],
xlab="Number of cyclical genes",
ylab="Proportion of Seurate genes",
main = "Seurat genes in top 260")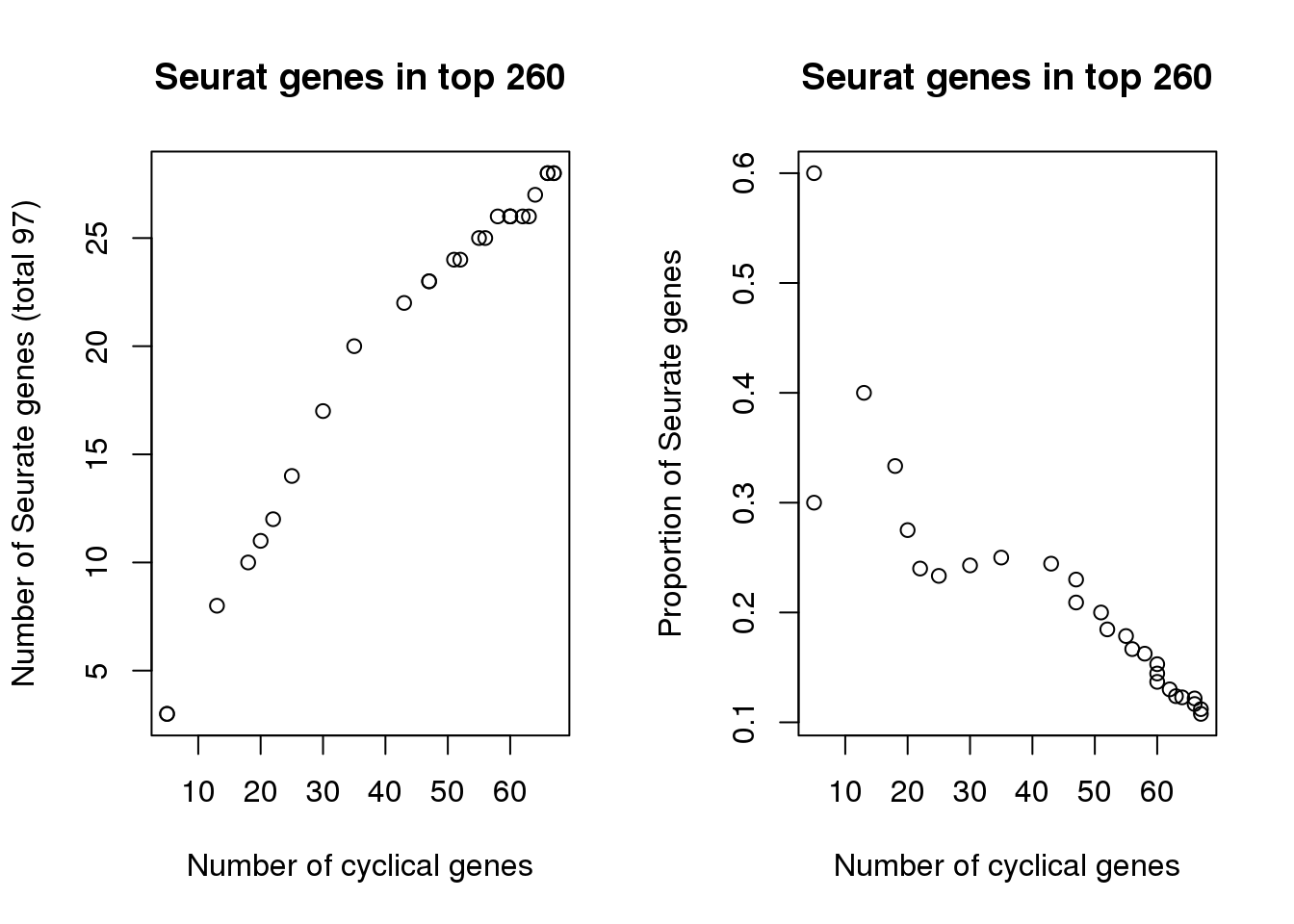
library(gplots)
venn(list(seurat=unlist(seurat.genes),
peco=genes_list_symbols[[27]]$hgnc_symbol))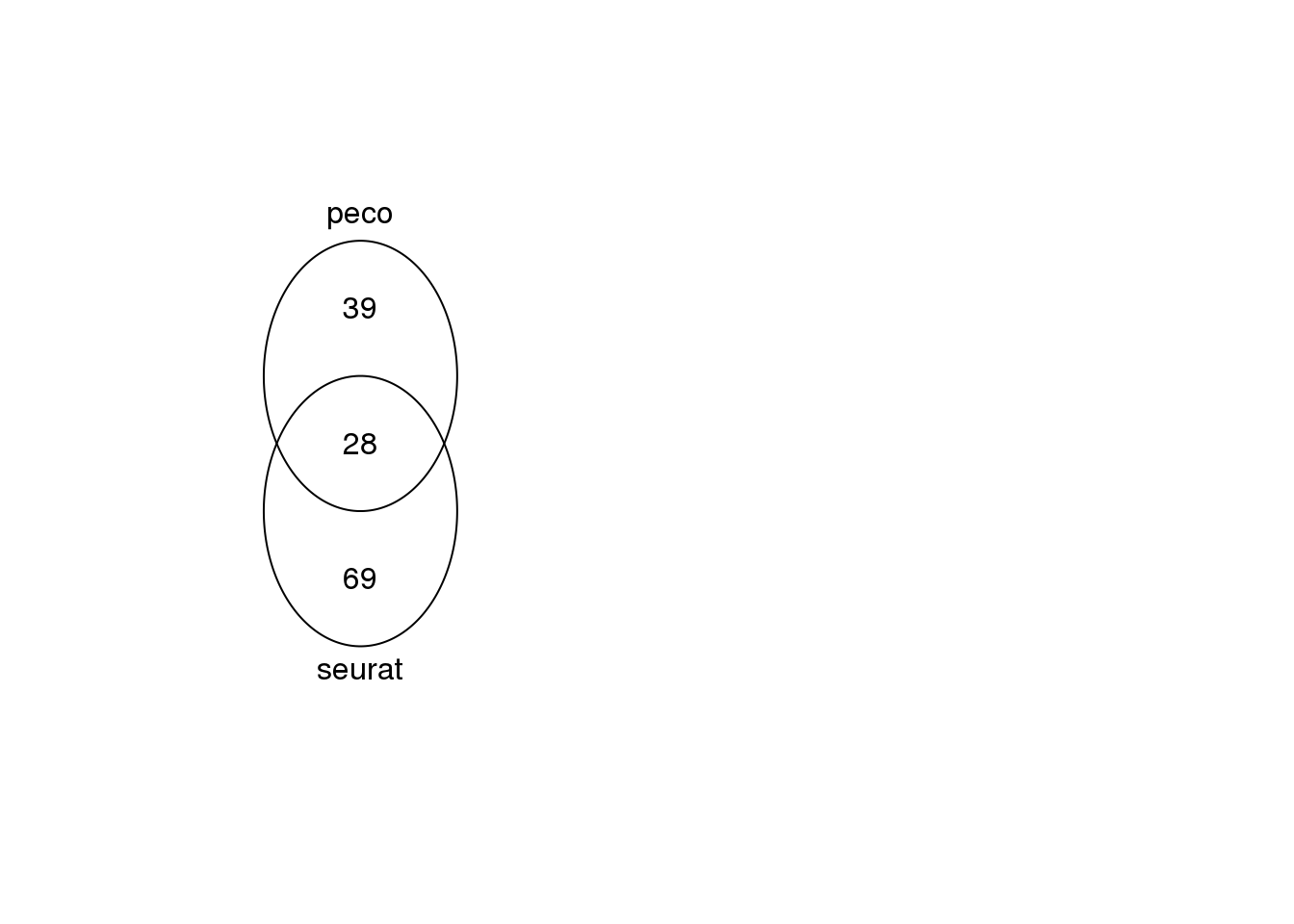
save to output table
write.table(genes_list_symbols[[27]]$hgnc_symbol,
file = "../output/method-train-classifiers-genes.Rmd/topgenes.txt",
row.names=F,
col.names=F, quote=F)
write.table(genes_list_symbols[[length(genes_list_symbols)]][,1],
file = "../output/method-train-classifiers-genes.Rmd/allgenes.txt",
row.names=F,
col.names=F, quote=F)Session information
sessionInfo()R version 3.4.3 (2017-11-30)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] gplots_3.0.1
loaded via a namespace (and not attached):
[1] Rcpp_0.12.17 gtools_3.5.0 digest_0.6.15
[4] rprojroot_1.3-2 bitops_1.0-6 backports_1.1.2
[7] git2r_0.21.0 magrittr_1.5 evaluate_0.10.1
[10] KernSmooth_2.23-15 stringi_1.1.6 gdata_2.18.0
[13] rmarkdown_1.10 tools_3.4.3 stringr_1.2.0
[16] yaml_2.1.16 compiler_3.4.3 caTools_1.17.1
[19] htmltools_0.3.6 knitr_1.20 This R Markdown site was created with workflowr