Subsampling Simulations for ALKATI
Haider Inam
1/31/2019
Last updated: 2019-02-11
workflowr checks: (Click a bullet for more information)-
✖ R Markdown file: uncommitted changes
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can runwflow_publishto commit the R Markdown file and build the HTML. -
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(20190211)The command
set.seed(20190211)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 5d432fb
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .Rproj.user/ Untracked files: Untracked: code/alldata_compiler.R Untracked: code/contab_maker.R Untracked: code/mut_excl_genes_datapoints.R Untracked: code/mut_excl_genes_generator.R Untracked: code/quadratic_solver.R Untracked: code/simresults_generator.R Untracked: data/All_Data_V2.csv Untracked: output/alkati_mtn_pval_fig2B.pdf Unstaged changes: Modified: analysis/alkati_subsampling_simulations.Rmd
Expand here to see past versions:
P-value distribution plots for Figure 1C:
nsubsamples=12 # maybe this can be removed and instead calculated later.
nsims<-100 #
#Positive control 1
nameposctrl1<-'BRAF'
#Positive control 1
nameposctrl2<-'NRAS'
#Oncogene in Question
namegene<-'ATI'
#Mutation Boolean (Y or N)
mtn<-'N'
#Name Mutation for Positive Ctrl 1
nameposctrl1mt<-'V600E'
#Name of Mutation for Positive Ctrl 2
nameposctrl2mt<-'Q61L'
alldata=read.csv("data/All_Data_V2.csv",sep=",",header=T,stringsAsFactors=F)
nexperiments=7
alldata_comp=alldata_compiler(alldata,nameposctrl1,nameposctrl2,namegene,mtn,"N/A","N/A")[[2]]
genex_replication_prop=alldata_compiler(alldata,nameposctrl1,nameposctrl2,namegene,mtn,"N/A","N/A")[[1]]
simresults_comb=data.frame()
for(subsample_number in c(1:12)){
nsubsamples=subsample_number
simresults=simresults_generator(alldata_comp,7)
simresults_comb=rbind(simresults_comb,simresults) ##iterative rbind this is not the most efficient way to do this
}simresults_concat=simresults_comb%>%
filter(exp_num%in%c(4))
# simresults_concat=simresults_comb
ggplot(simresults_concat,aes(x=factor(subsample_size),y=log10(p_val)))+
geom_boxplot(aes(fill=factor(exp_num)))+
cleanup+
guides(fill=F)+
scale_y_continuous(name="log(P-Value)")+
scale_x_discrete(name="Subsample size")+
# scale_color_manual(values="#E78AC3")+
theme(plot.title = element_text(hjust=.5),
text = element_text(size=26,face="bold"),
axis.title = element_text(face="bold",size="26",color="black"),
axis.text=element_text(face="bold",size="24",color="black"))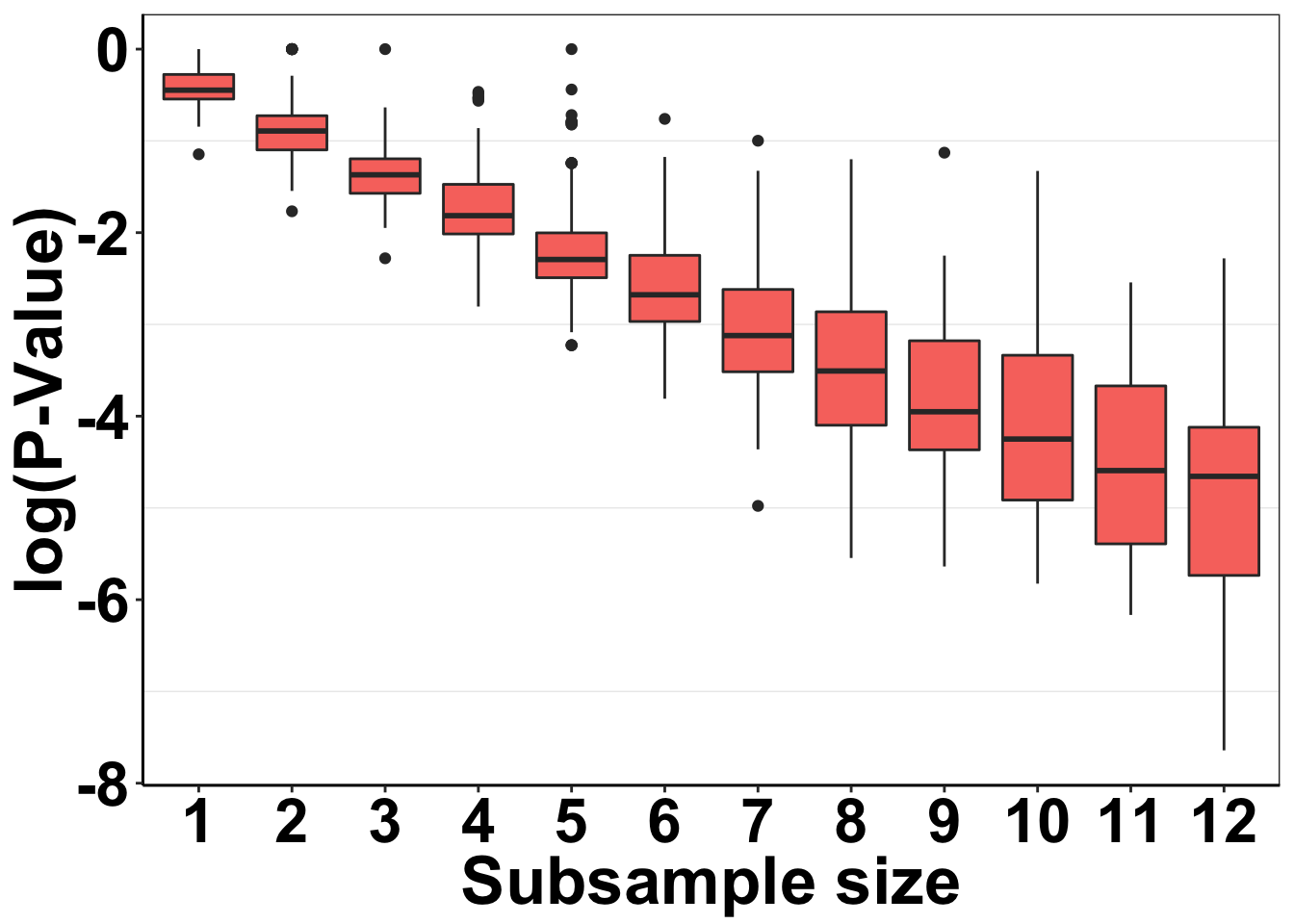
Expand here to see past versions of unnamed-chunk-2-1.png:
| Version | Author | Date |
|---|---|---|
| 5d432fb | haiderinam | 2019-02-11 |
# ggsave("output/ alkati_subsamplesize_pval_fig1c.pdf",width = 10,height = 10,units = "in",useDingbats=F)Adding simulations for comparisons with mutations:
nsubsamples=12 # maybe this can be removed and instead calculated later.
nsims<-100 #
#Positive control 1
nameposctrl1<-'BRAF'
#Positive control 1
nameposctrl2<-'NRAS'
#Oncogene in Question
namegene<-'ATI'
#Mutation Boolean (Y or N)
mtn<-'Y'
#Name Mutation for Positive Ctrl 1
nameposctrl1mt<-'V600E'
#Name of Mutation for Positive Ctrl 2
nameposctrl2mt<-'Q61L'
alldata=read.csv("data/All_Data_V2.csv",sep=",",header=T,stringsAsFactors=F)
nexperiments=7
###For mutation
alldata_comp=alldata_compiler(alldata,nameposctrl1,nameposctrl2,namegene,mtn,nameposctrl1mt,nameposctrl2mt)[[2]]
genex_replication_prop=alldata_compiler(alldata,nameposctrl1,nameposctrl2,namegene,mtn,nameposctrl1mt,nameposctrl2mt)[[1]]
simresults=simresults_generator(alldata_comp,7)
simresults$mtn='Y'
####For no mutation
mtn='N'
alldata_comp=alldata_compiler(alldata,nameposctrl1,nameposctrl2,namegene,mtn,"N/A","N/A")[[2]]
genex_replication_prop=alldata_compiler(alldata,nameposctrl1,nameposctrl2,namegene,mtn,"N/A","N/A")[[1]]
simresults_nomtn=simresults_generator(alldata_comp,7)
simresults_nomtn$mtn='N'
simresults=rbind(simresults,simresults_nomtn)Generating the P-value plots for Figure 2. Will show ati vs braf, ati vs nras, for with-muts and without-muts
simresults[simresults$exp_num==1,]$exp_name="BRAF & ALKATI"
simresults[simresults$exp_num==3,]$exp_name="NRAS & ALKATI"
simresults[simresults$exp_num==4,]$exp_name="BRAF & NRAS"
simresults$exp_name=factor(simresults$exp_name,levels=c("1","5","6","7","BRAF & ALKATI","NRAS & ALKATI","BRAF & NRAS"))
simresults$mtn_tag='N'
simresults[simresults$mtn=='Y',]$mtn_tag="Mutation-specific"
simresults[simresults$mtn=='N',]$mtn_tag="Non mutation-specific"
simresults$mtn_tag=factor(simresults$mtn_tag,levels=c("Non mutation-specific","Mutation-specific"))
simresults_concat=simresults%>%
filter(exp_num==c(1,3,4))Warning in exp_num == c(1, 3, 4): longer object length is not a multiple of
shorter object lengthggplot(simresults_concat,aes(x=factor(exp_name),y=log10(p_val)))+
geom_boxplot(aes(fill=factor(exp_name)))+
facet_wrap(~factor(mtn_tag))+
cleanup+
guides(fill=F)+
scale_y_continuous(name="log(P-Value)")+
scale_x_discrete(name="Gene Pair")+
scale_fill_brewer(palette = "Set2",name="Gene Pair")+
theme(plot.title = element_text(hjust=.5),
text = element_text(size=26,face="bold"),
axis.title = element_text(face="bold",size="26",color="black"),
axis.text=element_text(face="bold",size="20",color="black"))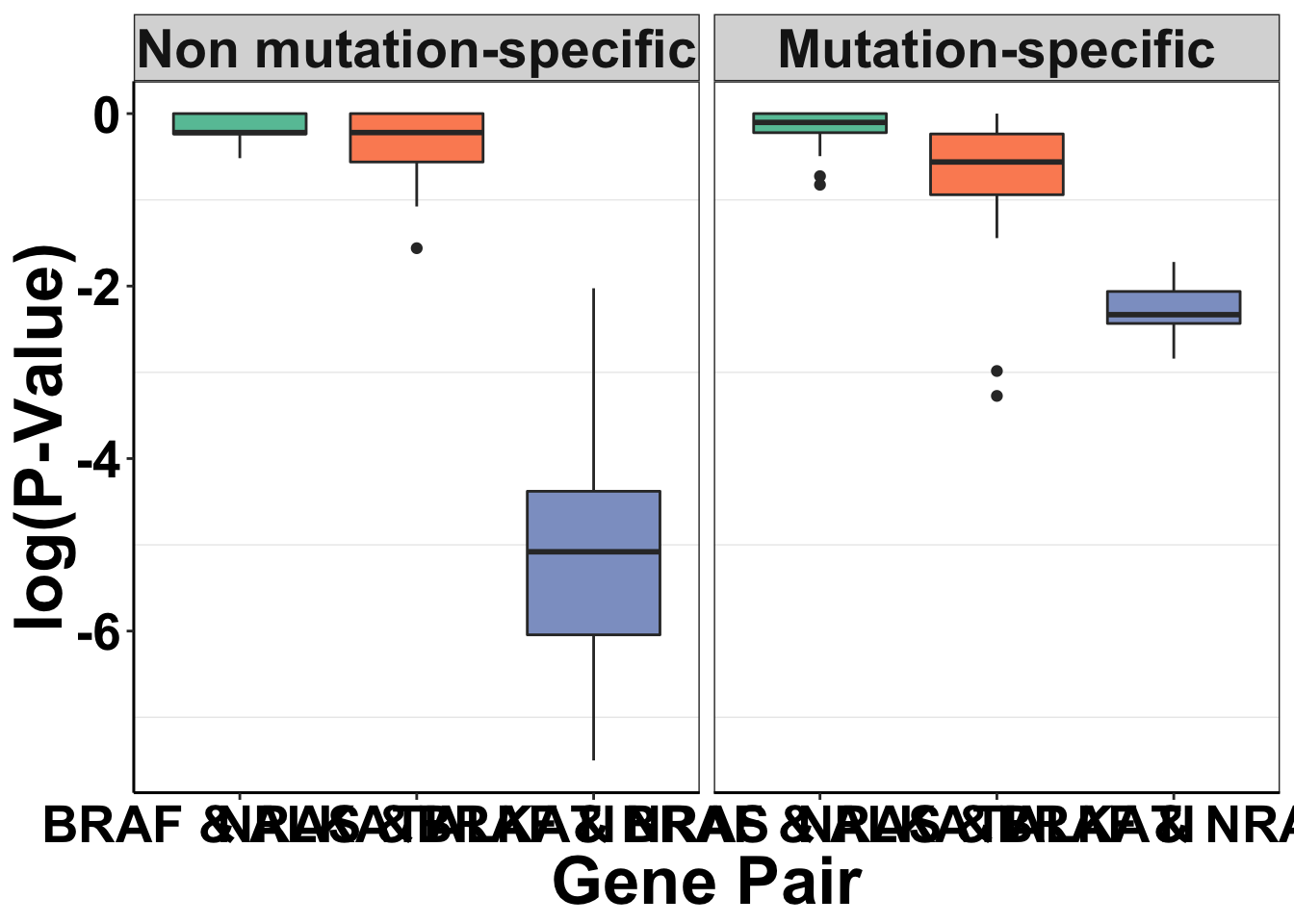
Expand here to see past versions of unnamed-chunk-4-1.png:
| Version | Author | Date |
|---|---|---|
| 5d432fb | haiderinam | 2019-02-11 |
ggsave("output/alkati_mtn_pval_fig2B.pdf",width = 16,height = 10,units = "in",useDingbats=F)Session information
sessionInfo()R version 3.5.2 (2018-12-20)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Mojave 10.14.3
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel grid stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] bindrcpp_0.2.2 ggsignif_0.4.0 usethis_1.4.0
[4] devtools_2.0.1 RColorBrewer_1.1-2 reshape2_1.4.3
[7] ggplot2_3.1.0 doParallel_1.0.14 iterators_1.0.10
[10] foreach_1.4.4 dplyr_0.7.8 VennDiagram_1.6.20
[13] futile.logger_1.4.3 workflowr_1.1.1 tictoc_1.0
[16] knitr_1.21
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 xfun_0.4 remotes_2.0.2
[4] purrr_0.3.0 colorspace_1.4-0 htmltools_0.3.6
[7] yaml_2.2.0 rlang_0.3.1 pkgbuild_1.0.2
[10] R.oo_1.22.0 pillar_1.3.1 glue_1.3.0
[13] withr_2.1.2 R.utils_2.7.0 sessioninfo_1.1.1
[16] lambda.r_1.2.3 bindr_0.1.1 plyr_1.8.4
[19] stringr_1.3.1 munsell_0.5.0 gtable_0.2.0
[22] R.methodsS3_1.7.1 codetools_0.2-16 evaluate_0.12
[25] memoise_1.1.0 labeling_0.3 callr_3.1.1
[28] ps_1.3.0 Rcpp_1.0.0 backports_1.1.3
[31] scales_1.0.0 formatR_1.5 desc_1.2.0
[34] pkgload_1.0.2 fs_1.2.6 digest_0.6.18
[37] stringi_1.2.4 processx_3.2.1 rprojroot_1.3-2
[40] cli_1.0.1 tools_3.5.2 magrittr_1.5
[43] lazyeval_0.2.1 tibble_2.0.1 futile.options_1.0.1
[46] crayon_1.3.4 whisker_0.3-2 pkgconfig_2.0.2
[49] prettyunits_1.0.2 assertthat_0.2.0 rmarkdown_1.11
[52] rstudioapi_0.9.0 R6_2.3.0 git2r_0.24.0
[55] compiler_3.5.2 This reproducible R Markdown analysis was created with workflowr 1.1.1