R Notebook
Last updated: 2018-10-30
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(20181026)The command
set.seed(20181026)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: f7080f1
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: docs/figure/ Untracked files: Untracked: code/out/ Untracked: code/run-alignment.R Untracked: plots/ Unstaged changes: Modified: code/regressout-cellcycle.R
Expand here to see past versions:
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | f7080f1 | PytrikFolkertsma | 2018-10-30 | wflow_publish(c(“analysis/10x-180504-general-analysis.Rmd”, |
Analysis of the 10x samples: - tSNE plots - Cell cycle regression - PCA - Alignment - Marker gene expression - tSNE colored on metadata
library(Seurat)Loading required package: ggplot2Loading required package: cowplot
Attaching package: 'cowplot'The following object is masked from 'package:ggplot2':
ggsaveLoading required package: Matrixlibrary(ggplot2)
library(dplyr)
Attaching package: 'dplyr'The following objects are masked from 'package:stats':
filter, lagThe following objects are masked from 'package:base':
intersect, setdiff, setequal, unionall10x <- readRDS('output/10x-180504')
all10x.ccregout <- readRDS('output/10x-180504-ccregout')
#all10x.aligned <- readRDS('../output/10x-180504-aligned')
#all10x.aligned.ccregout <- readRDS('../output/10x-180504-ccregout-aligned')QC Plots
VlnPlot(all10x, features.plot='nGene', group.by='sample_name', point.size.use=-1, x.lab.rot=T)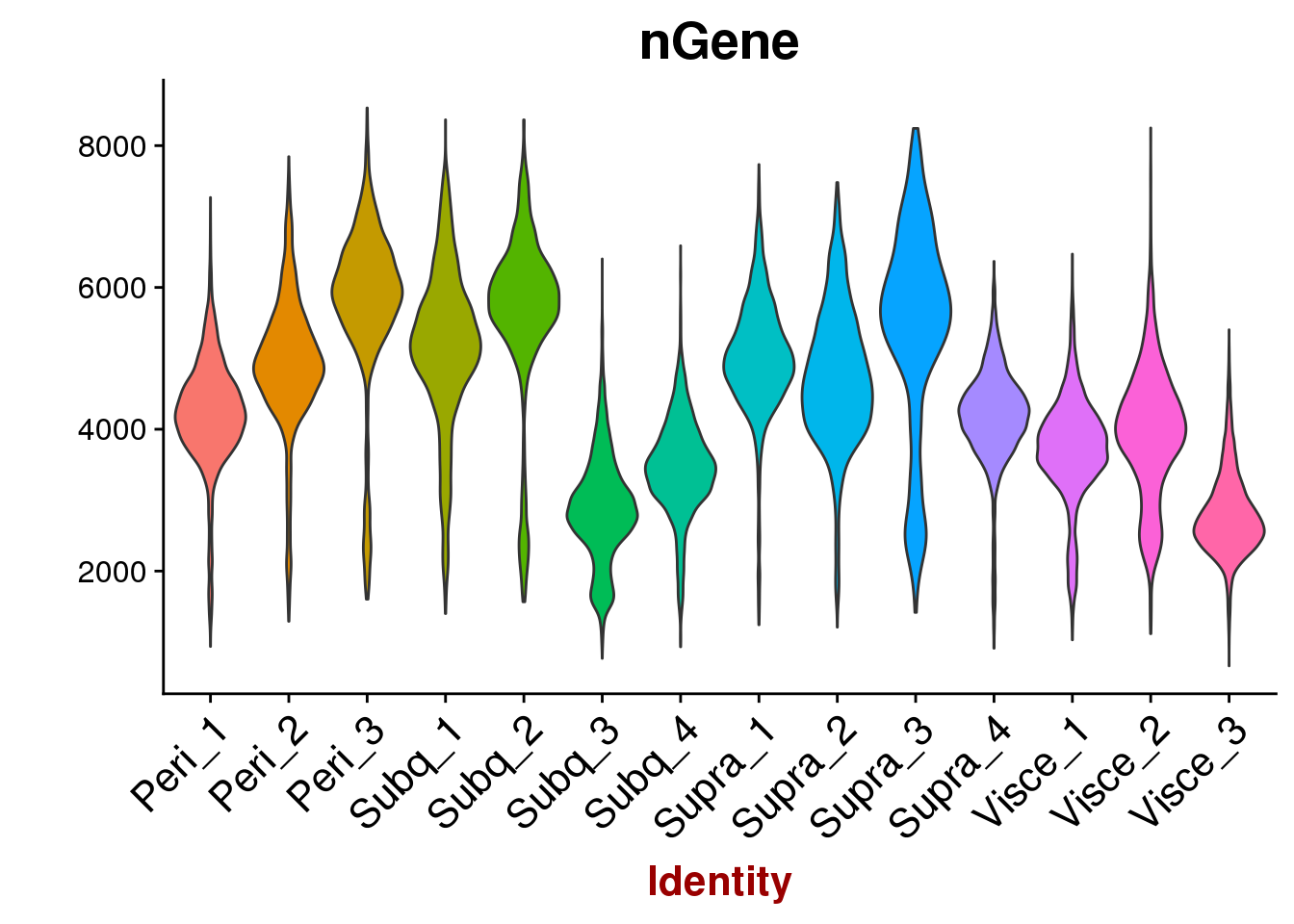
VlnPlot(all10x, features.plot='nUMI', group.by='sample_name', point.size.use=-1, x.lab.rot=T)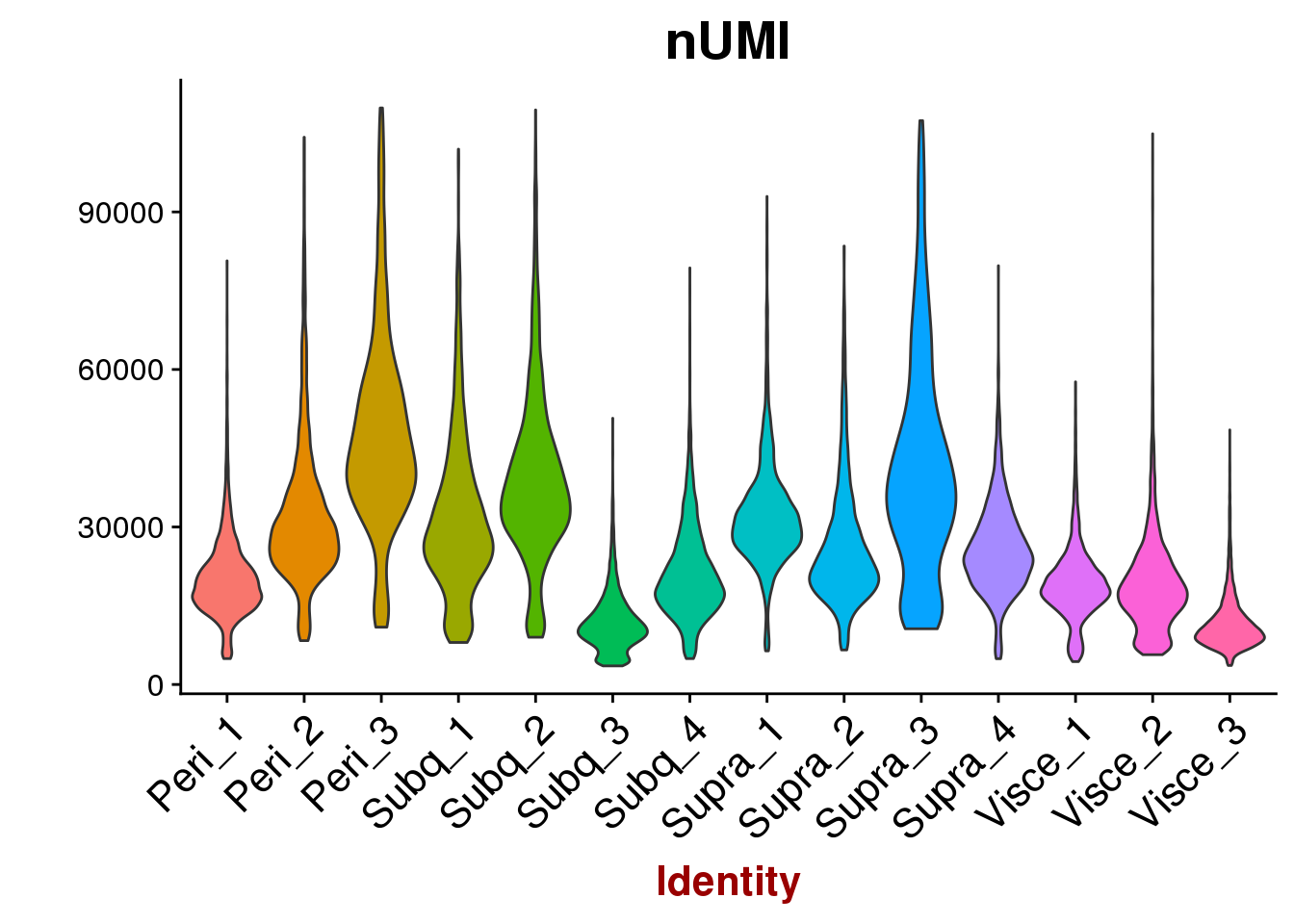
VlnPlot(all10x, features.plot='percent.mito', group.by='sample_name', point.size.use=-1, x.lab.rot=T)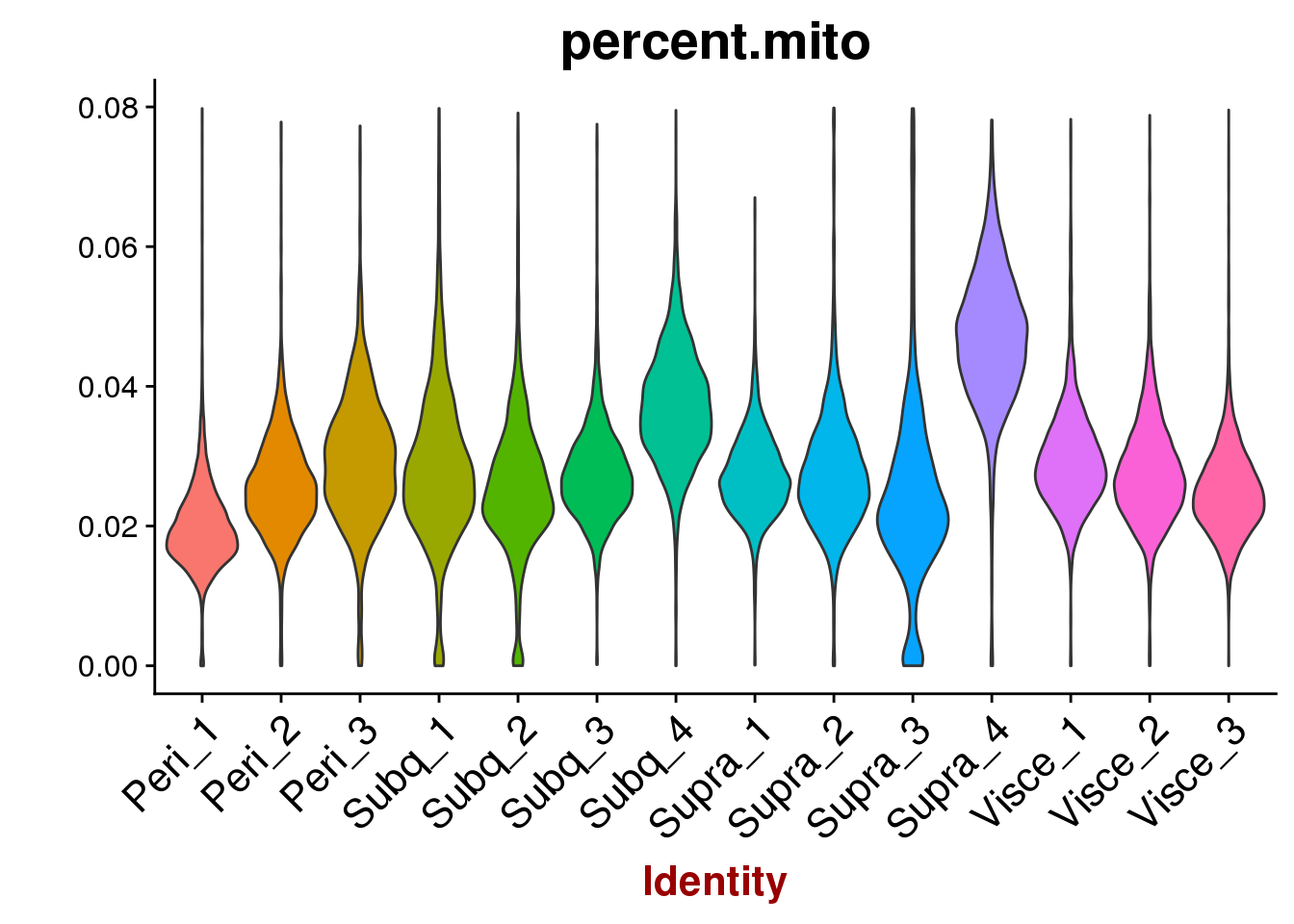
GenePlot(all10x, 'nUMI', 'nGene')
TSNE
Below are several tSNE plots of the 10x-180504 data. tSNE was performed on the first 15 principal components of the log-normalized scaled (nUMI and percent.mito regressed out) data.
Visceral and perirenal seem a bit mixed, and supraclavicular and subcutaneous too.
TSNEPlot(all10x, pt.size=0.1, group.by='sample_name', do.label=T)
tSNE plots of samples within their depot. Peri2 and Peri3 seem to overlap really well, as well as Supra1 and Supra2, and Visce1 and Visce3.
plot_grid(t1, t2, t3, t4)
tSNE colored on subtissue.
TSNEPlot(all10x, group.by='depot', pt.size=0.1)
tSNE colored by cell cycle phase.
TSNEPlot(all10x, group.by='Phase', pt.size=0.1)
Clusters
Some clustering with different resolutions. res=0.5
TSNEPlot(all10x, pt.size=0.1, group.by='res.0.5', do.label=T)
res=0.7
TSNEPlot(all10x, pt.size=0.1, group.by='res.0.7', do.label=T)
res=1
TSNEPlot(all10x, pt.size=0.1, group.by='res.1', do.label=T)
TSNEPlot(all10x, pt.size=0.1, group.by='sample_name', do.label=T)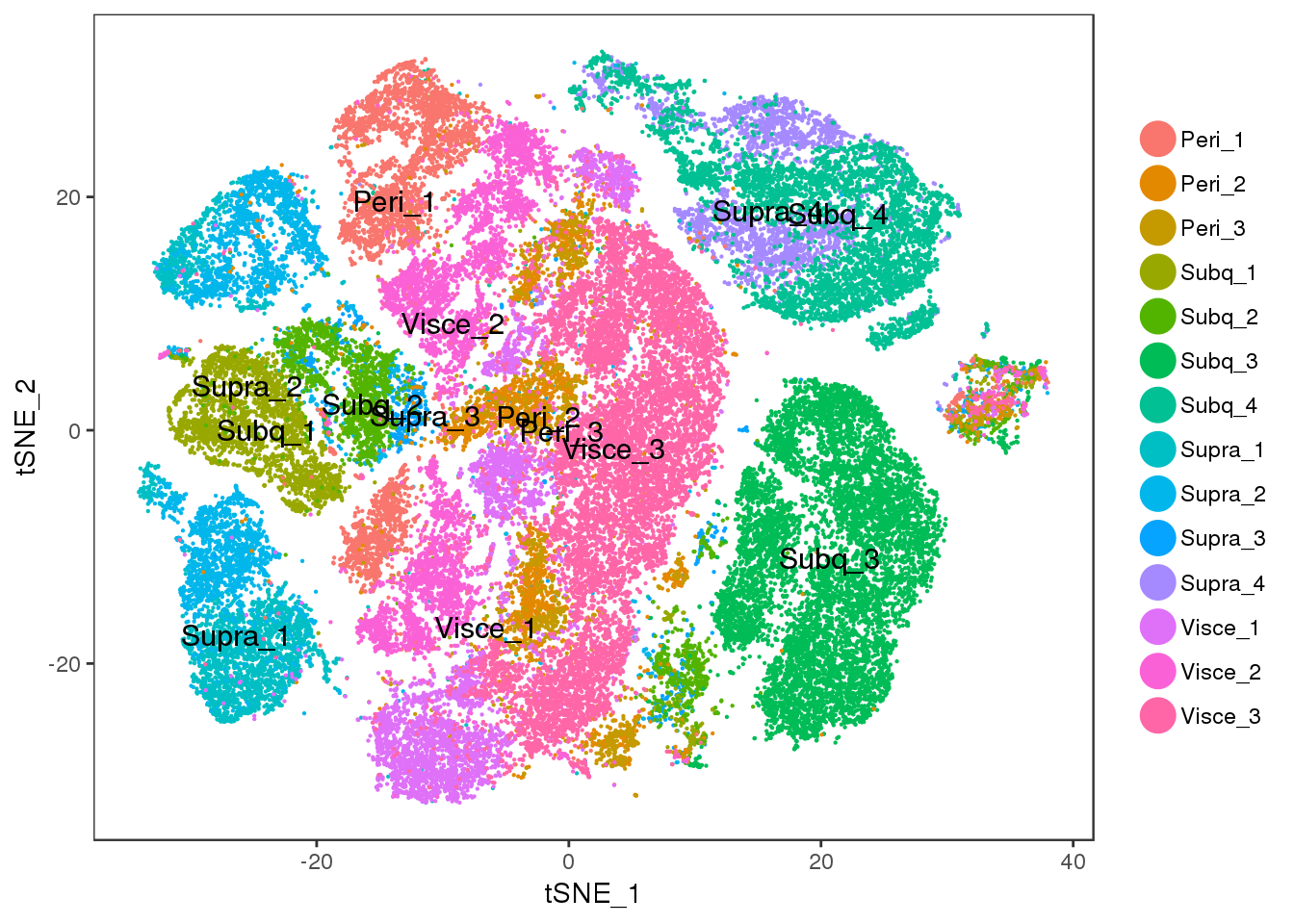
Cell cycle regression
T-SNE of the data with cell cycle effects regressed out. There does not seem to be a lot of structure within clusters now.
TSNEPlot(all10x.ccregout, pt.size=0.1, group.by='sample_name')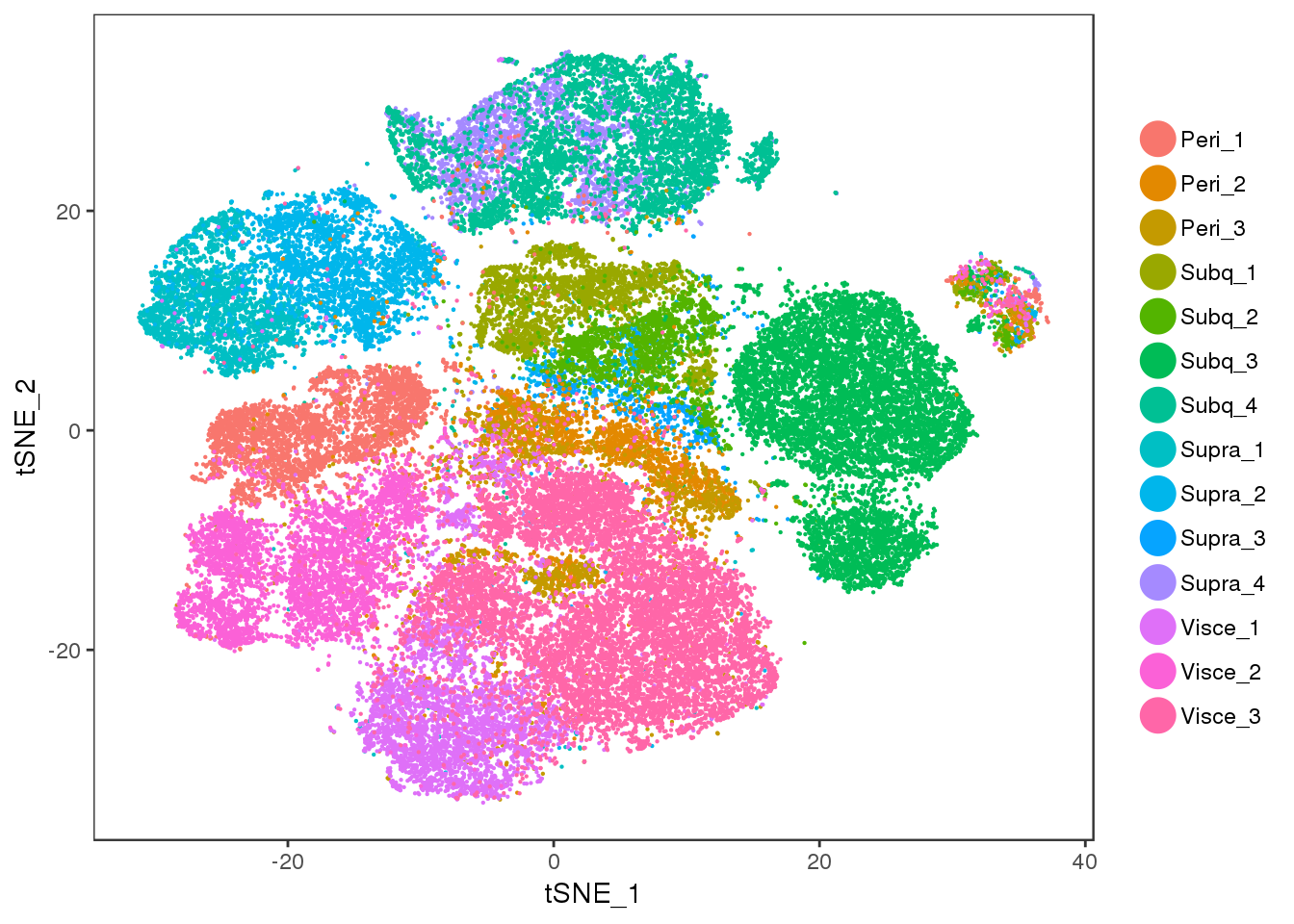
No cell cycle effect anymore.
TSNEPlot(all10x.ccregout, pt.size=0.1, group.by='Phase')
Subtissues
plot_grid(t1, t2, t3, t4)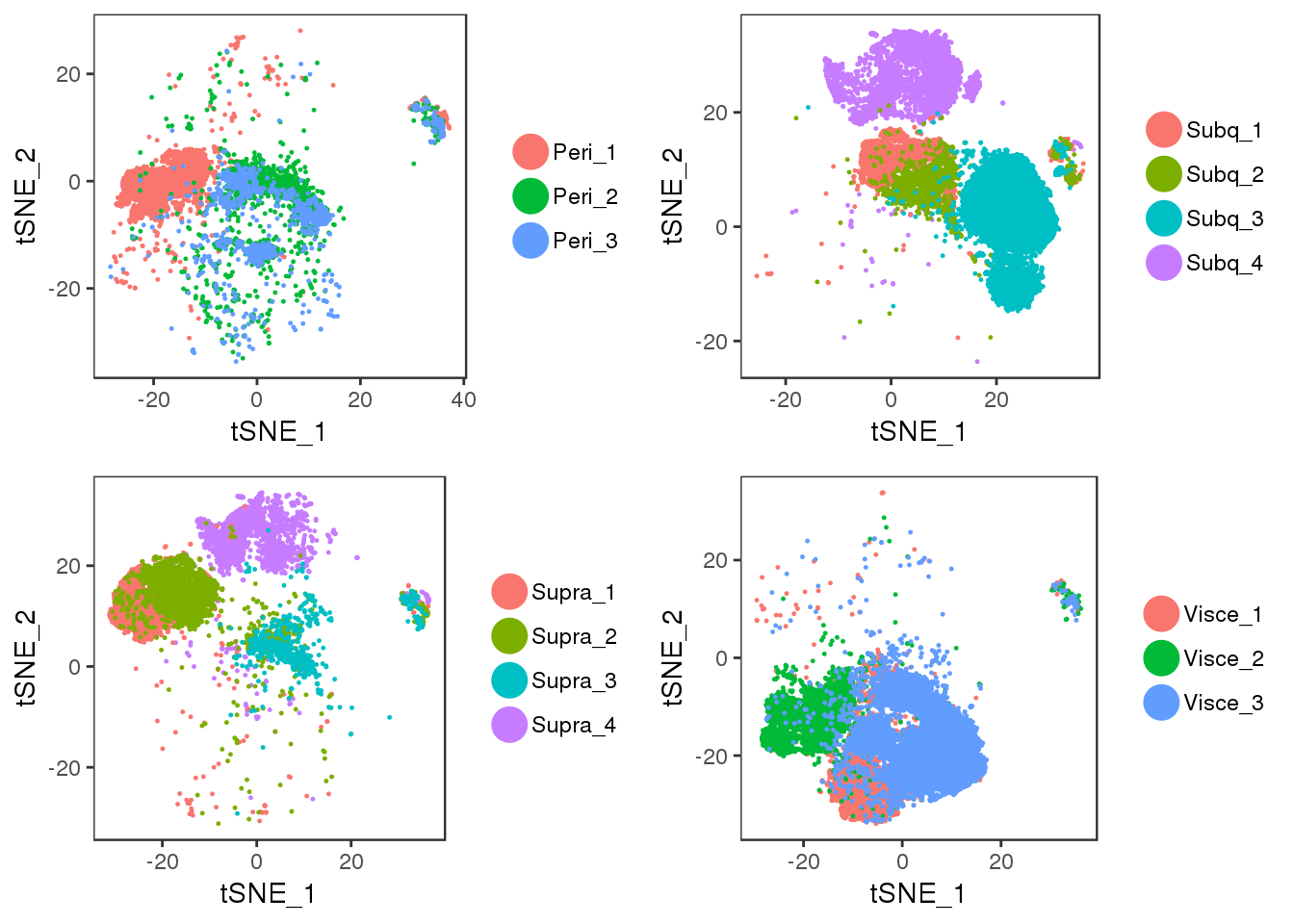
TSNEPlot(all10x.ccregout, pt.size=0.1, group.by='depot')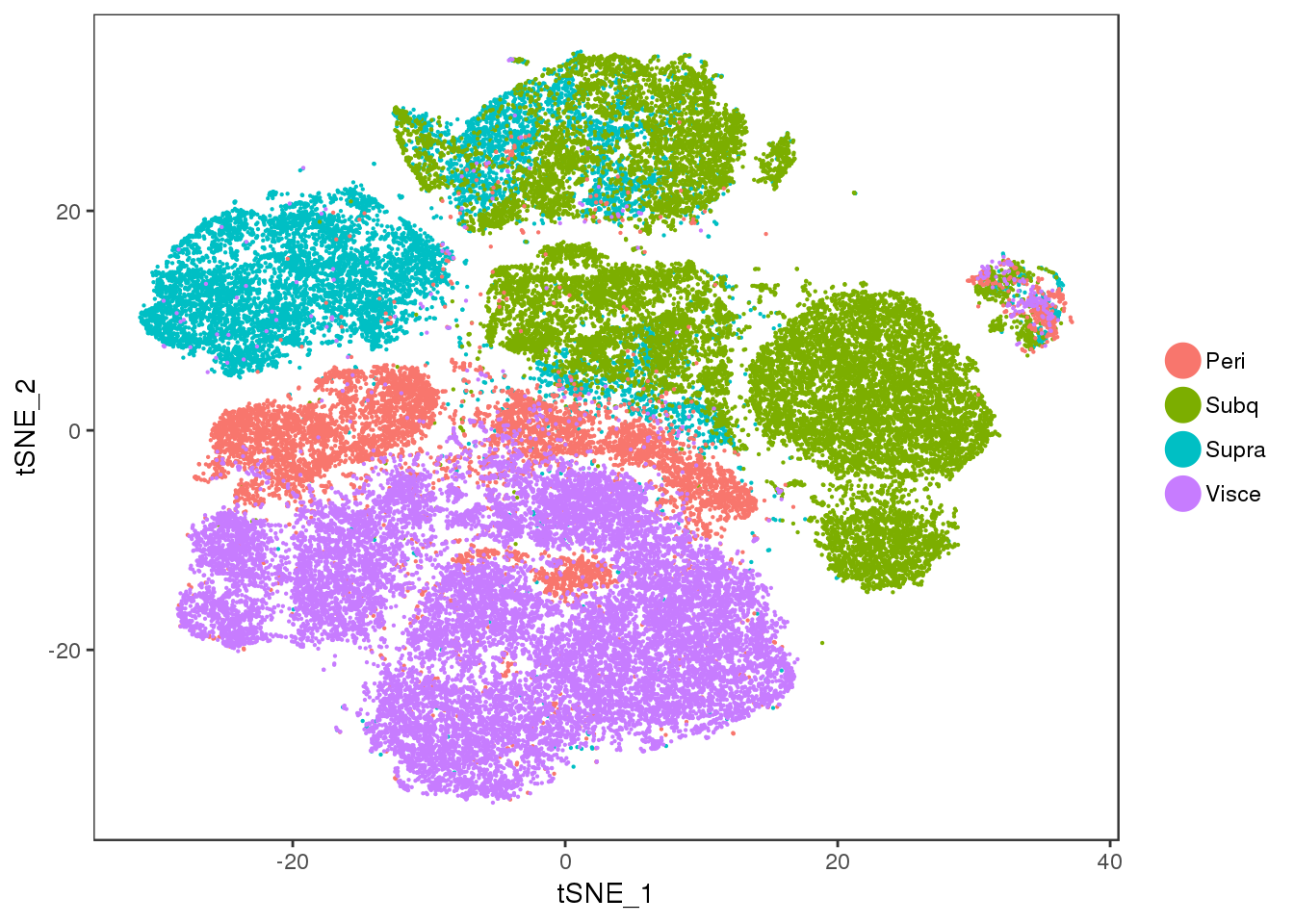
PCA
Some PCA plots. PC1 seems to capture cell cycle effects, and PC2 seems to capture some of the sample variability.
PCAPlot(all10x, group.by='Phase', pt.size=0.1)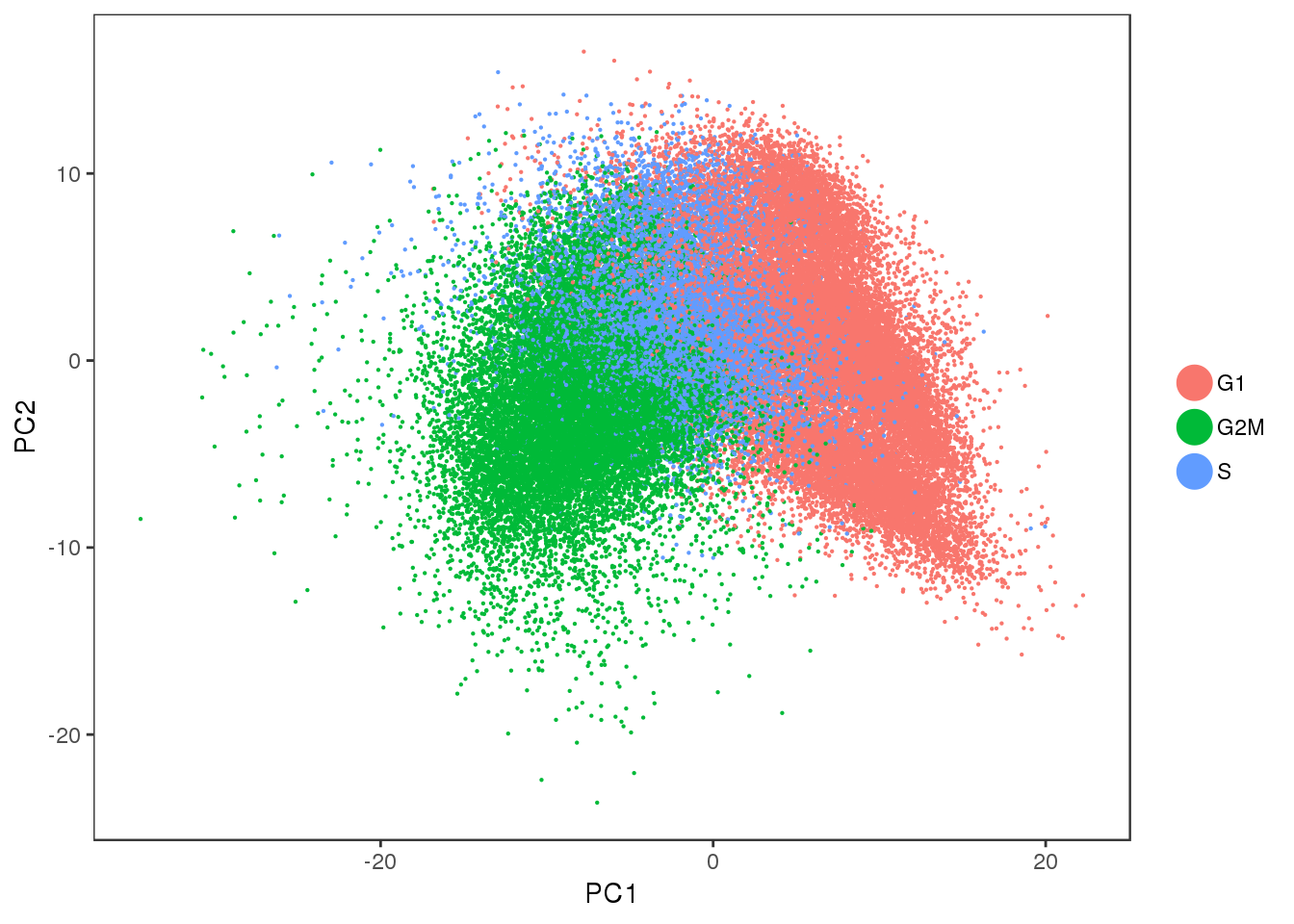
PCAPlot(all10x, group.by='sample_name', pt.size=0.1)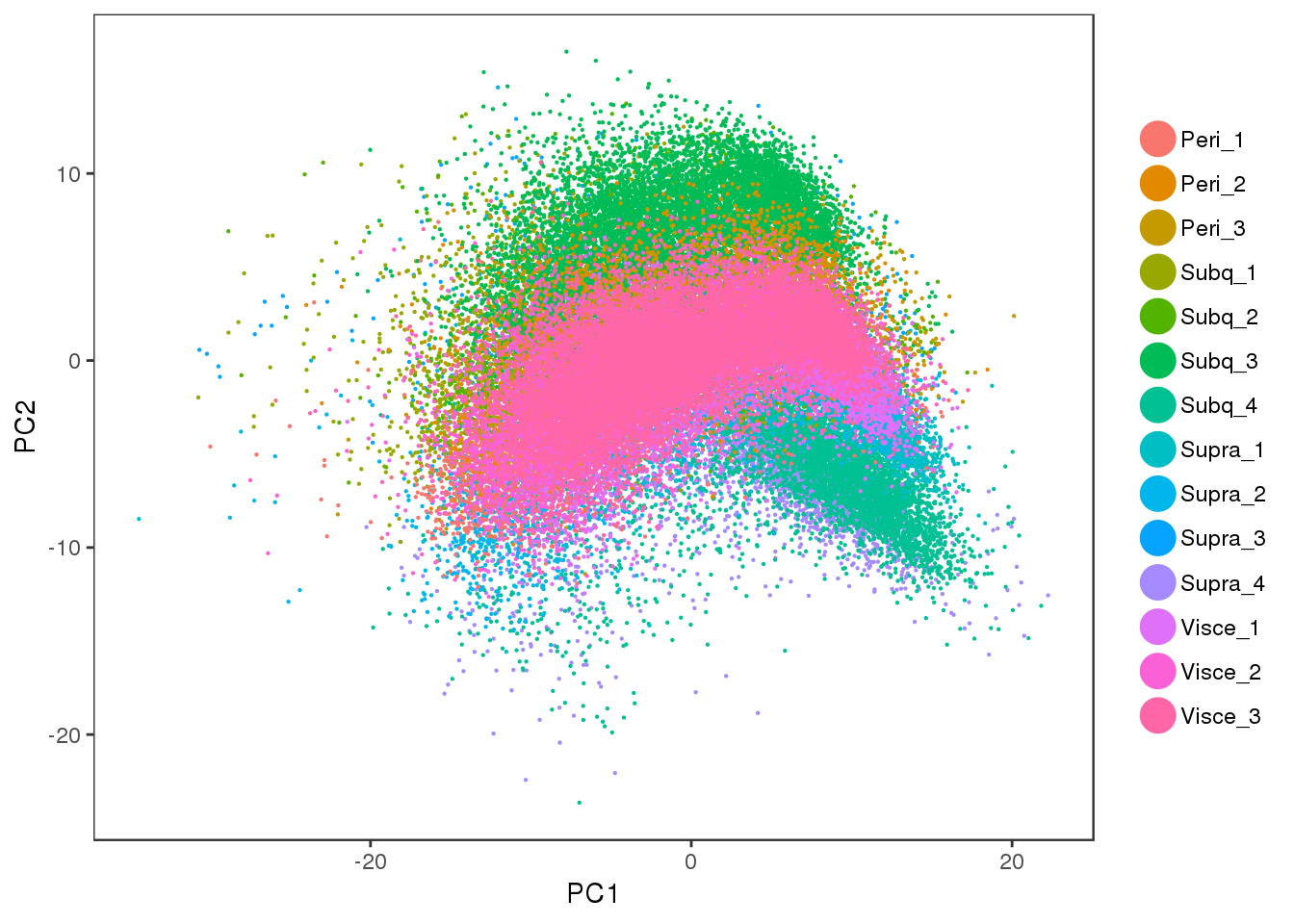
PCA plot of the cell cycle regressed out data. There is no cell cycle effect anymore.
PCAPlot(all10x.ccregout, group.by='Phase', pt.size=0.1)
PCAPlot(all10x.ccregout, group.by='sample_name', pt.size=0.1)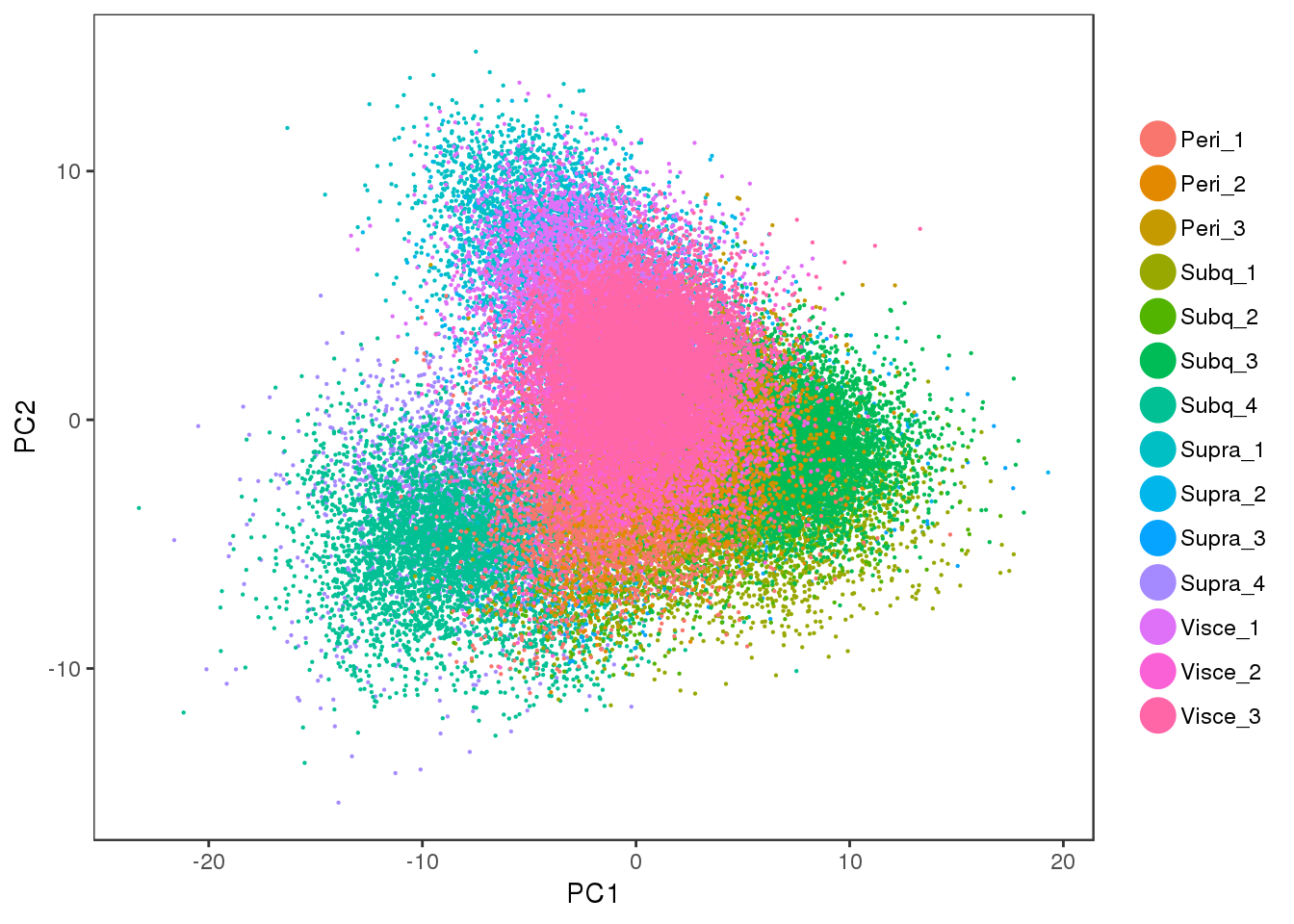
Alignment
Alignment of the data with and without cell cycle effects regressed out. Both were aligned on 30 subspaces, tSNE was performed on the first 15 CCs.
tSNE of the aligned data.
#TSNEPlot(all10x.aligned, group.by='sample_name', pt.size=0.1)tSNE of the aligned data coloured on cell cycle phase.
#TSNEPlot(all10x.aligned, group.by='Phase', pt.size=0.1)tSNE of the aligned data with cell cycle effects regressed out.
#TSNEPlot(all10x.aligned.ccregout, group.by='sample_name', pt.size=0.1)tSNE of the aligned data with cell cycle effects regressed out, colored by phase.
#TSNEPlot(all10x.aligned.ccregout, group.by='Phase', pt.size=0.1)tSNE of the aligned data with cell cycle effects regressed out, colored by subtissue
#TSNEPlot(all10x.aligned.ccregout, group.by='sample_name2', pt.size=0.1)Metadata plots
FeaturePlot(all10x, c("nGene"), cols.use = c("grey","blue"), no.legend=F)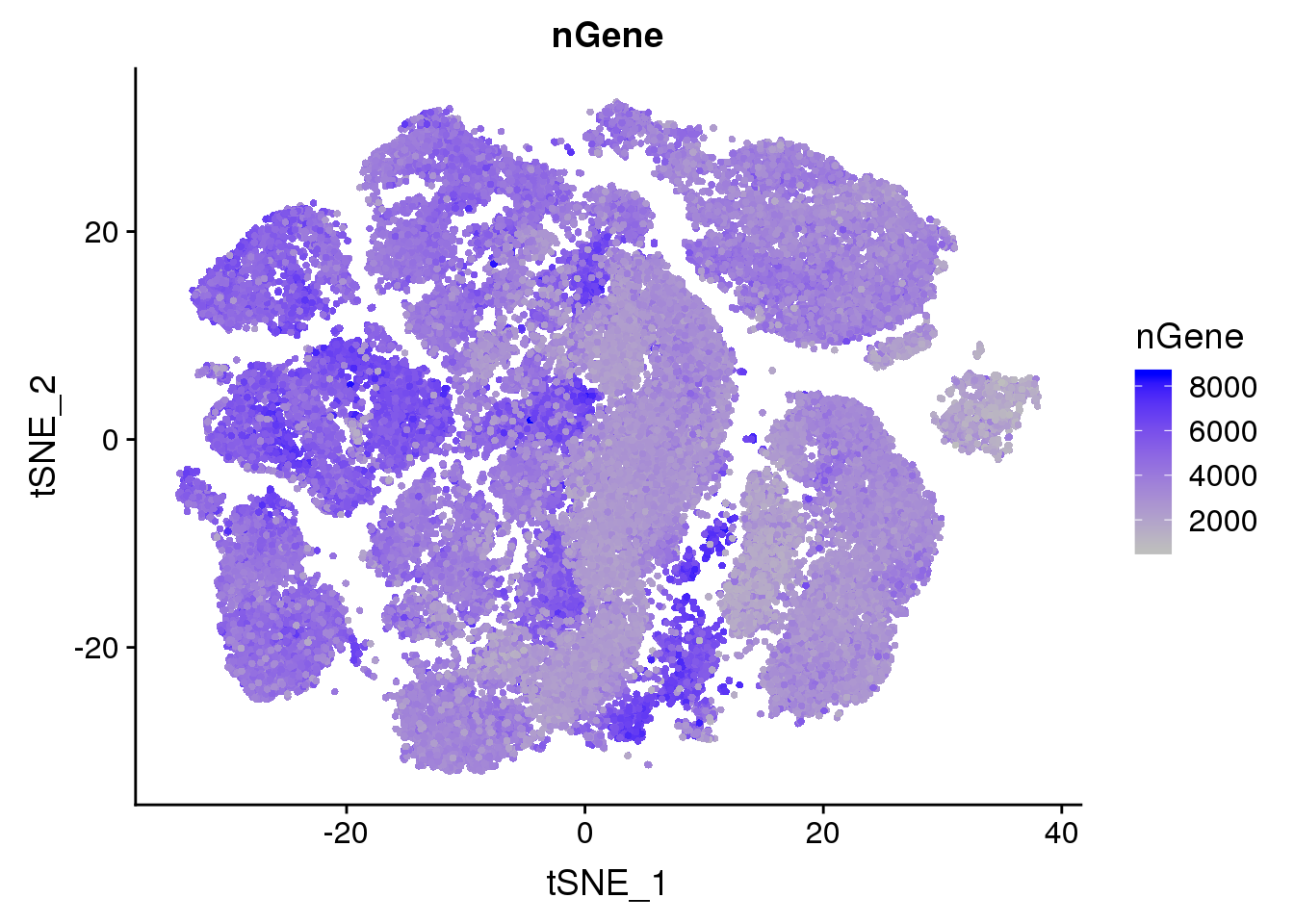
FeaturePlot(all10x, c("percent.mito"), cols.use = c("grey","blue"), no.legend=F)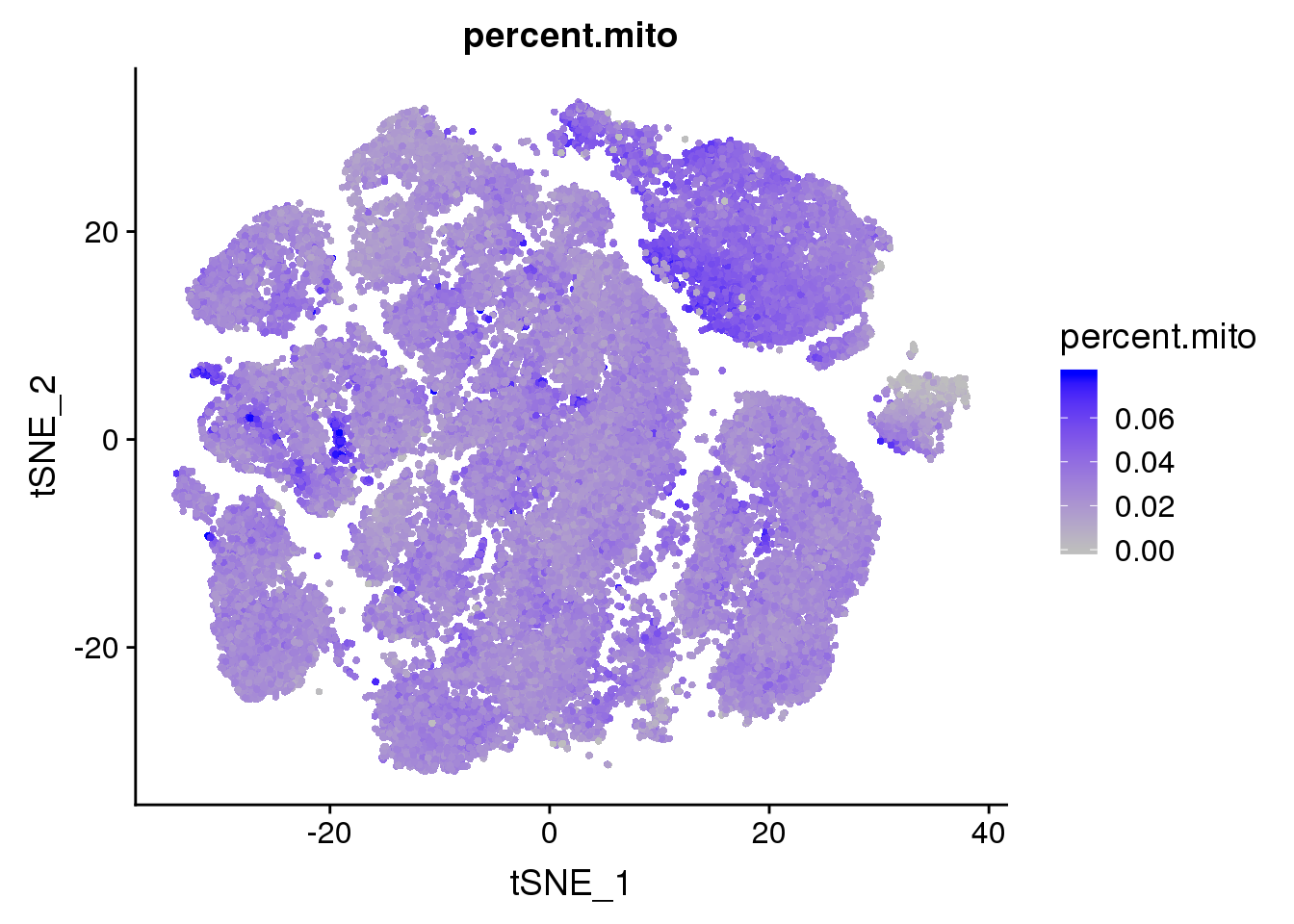
FeaturePlot(all10x, c("nUMI"), cols.use = c("grey","blue"), no.legend=F)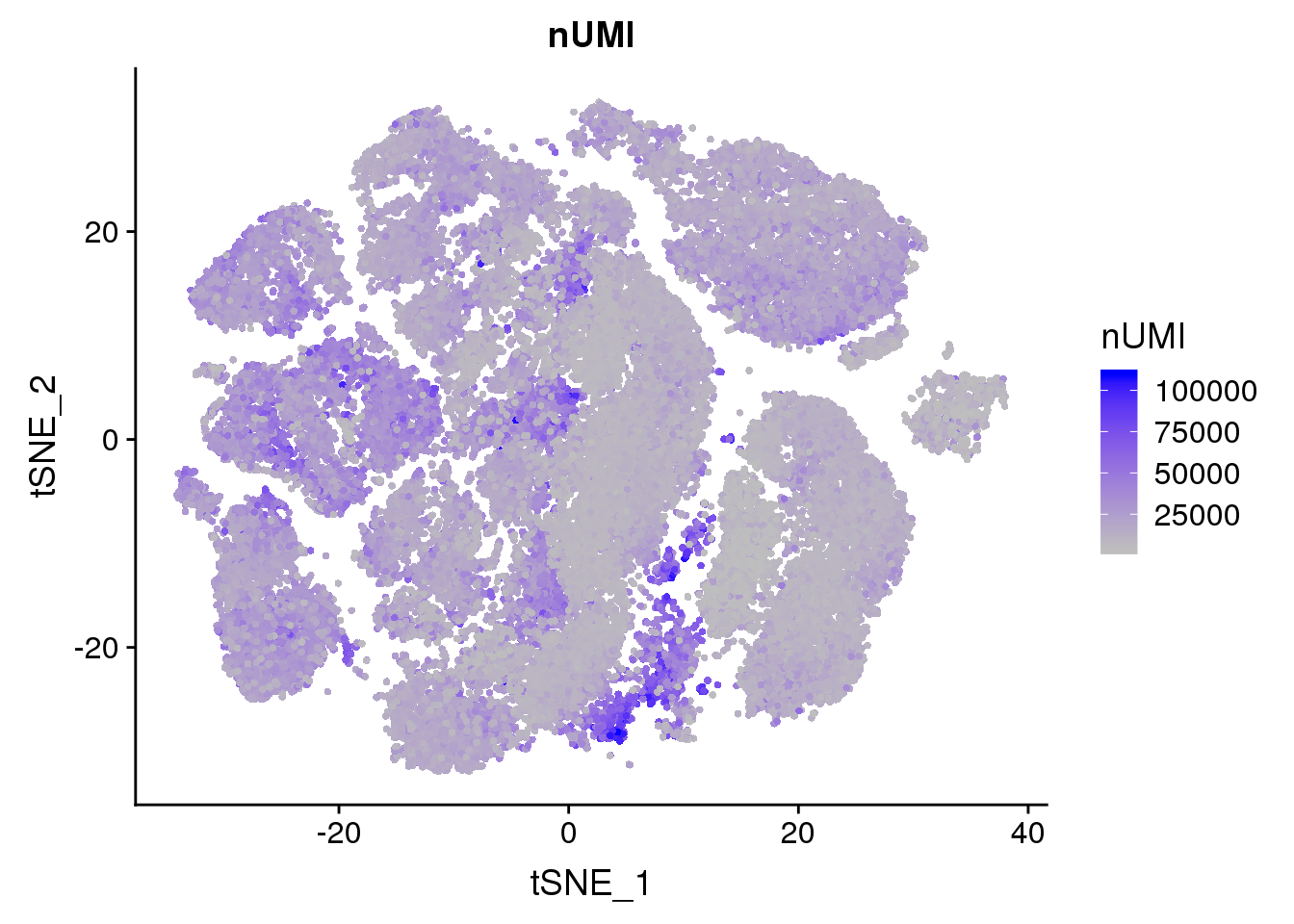
Diff
TSNEPlot(all10x, group.by='diff', pt.size=0.1)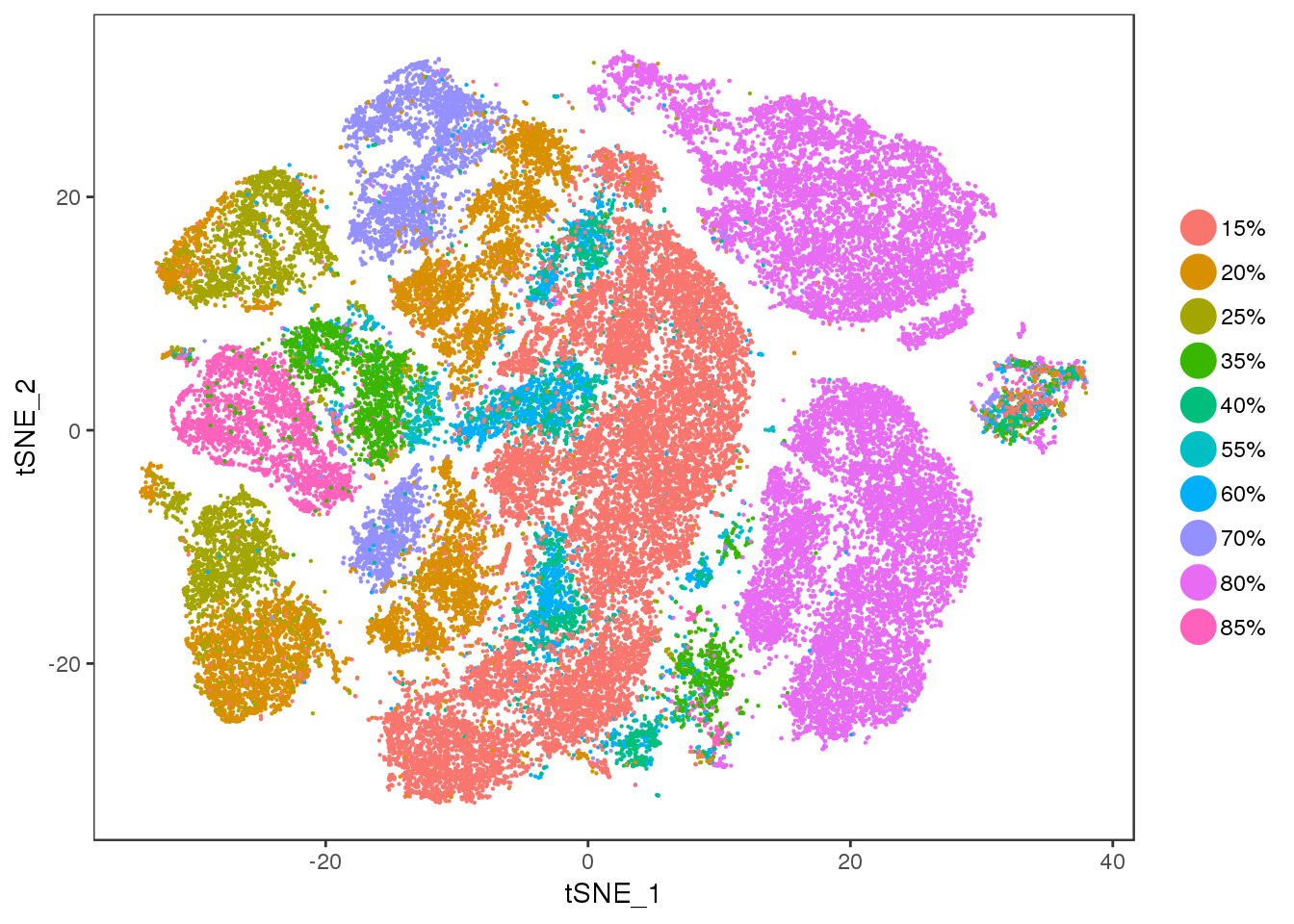
all10x@meta.data['diff_int'] <- unlist(lapply(as.vector(unlist(all10x@meta.data$diff)), function(x){return(strtoi(strsplit(x, '%')))}))
FeaturePlot(all10x, features.plot='diff_int', cols.use=c('gray', 'blue'), no.legend=F)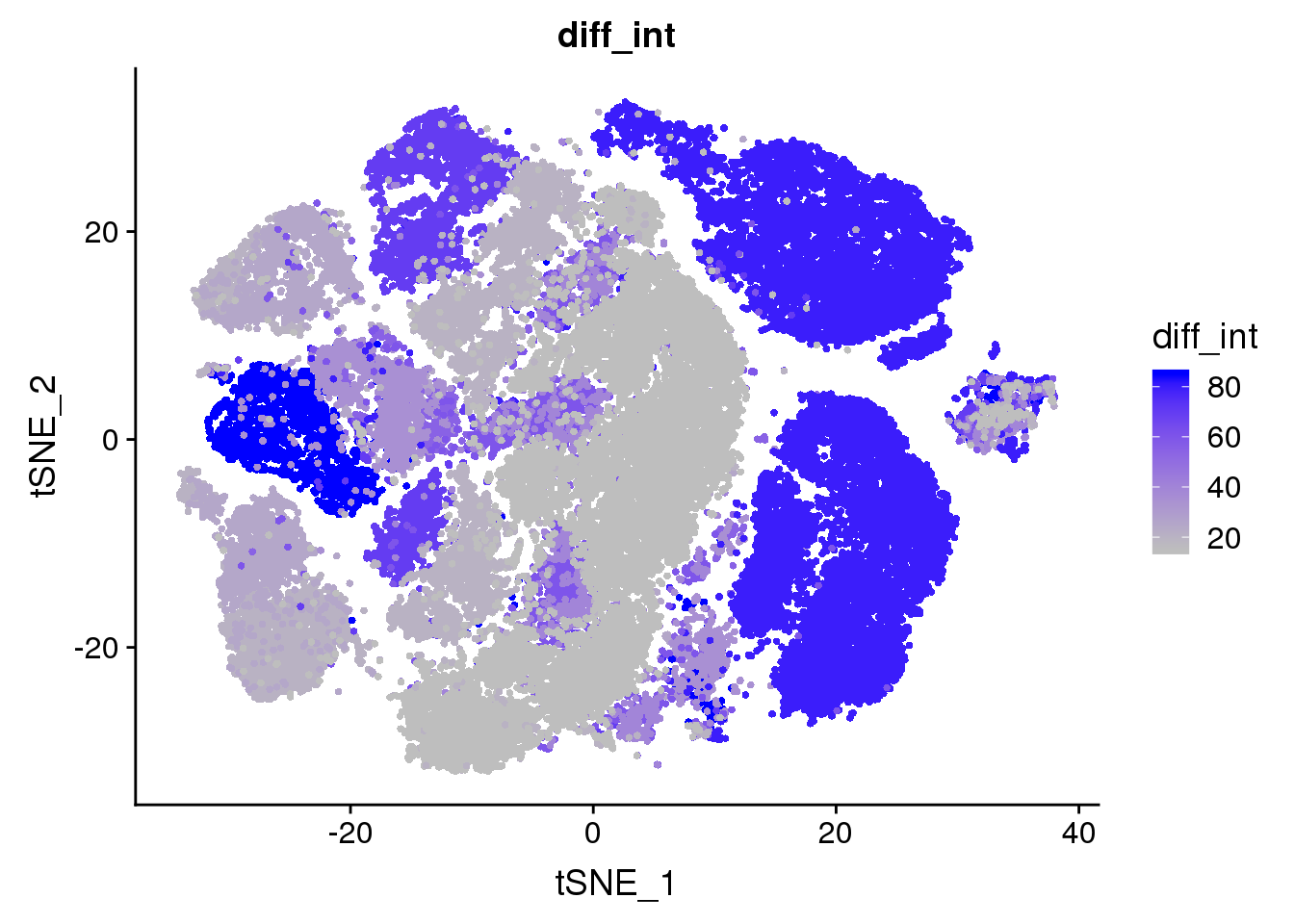
ucp1.ctrl
TSNEPlot(all10x, group.by='ucp1.ctrl', pt.size=0.1)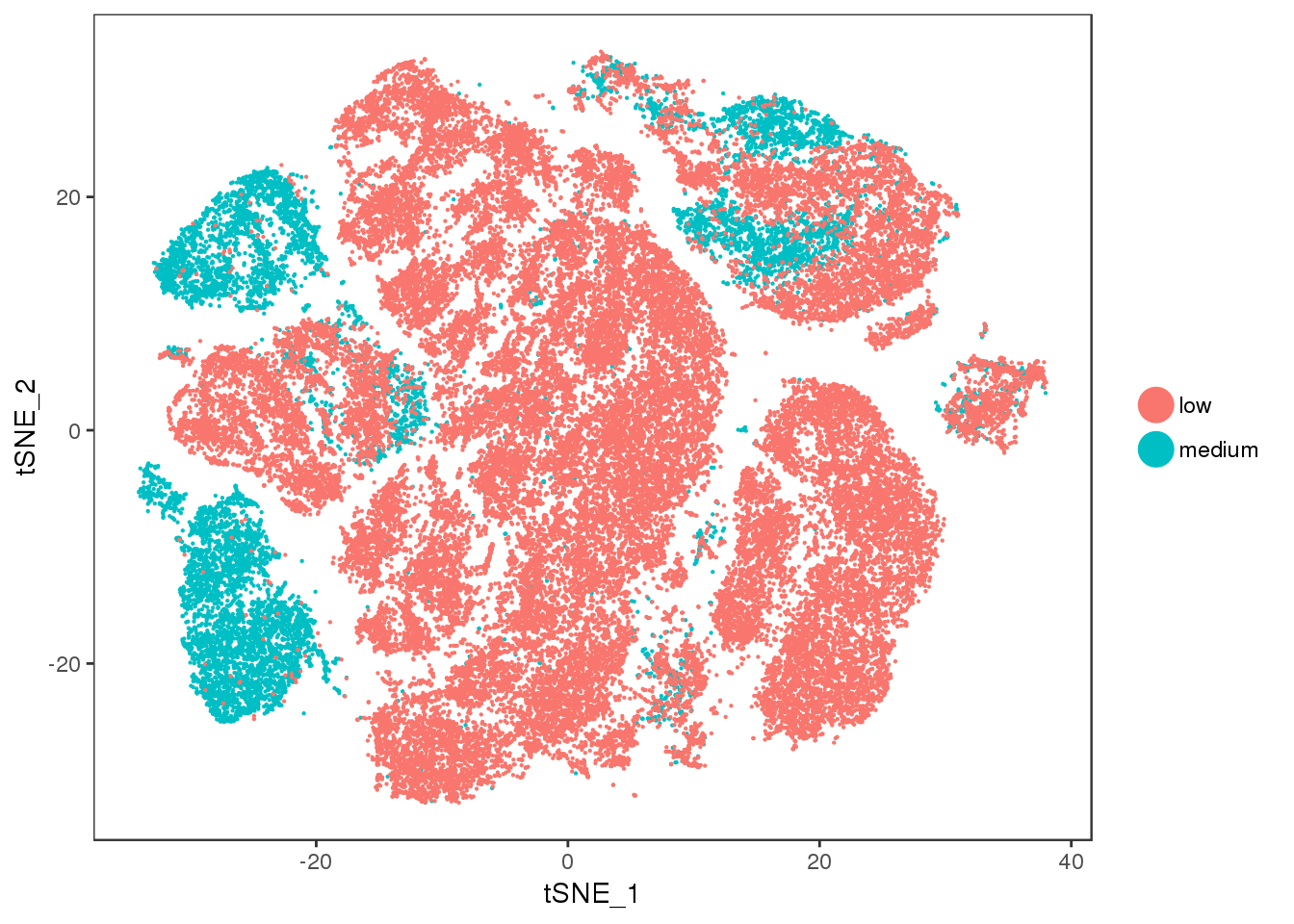
ucp1.ne
TSNEPlot(all10x, group.by='ucp1.ne', pt.size=0.1)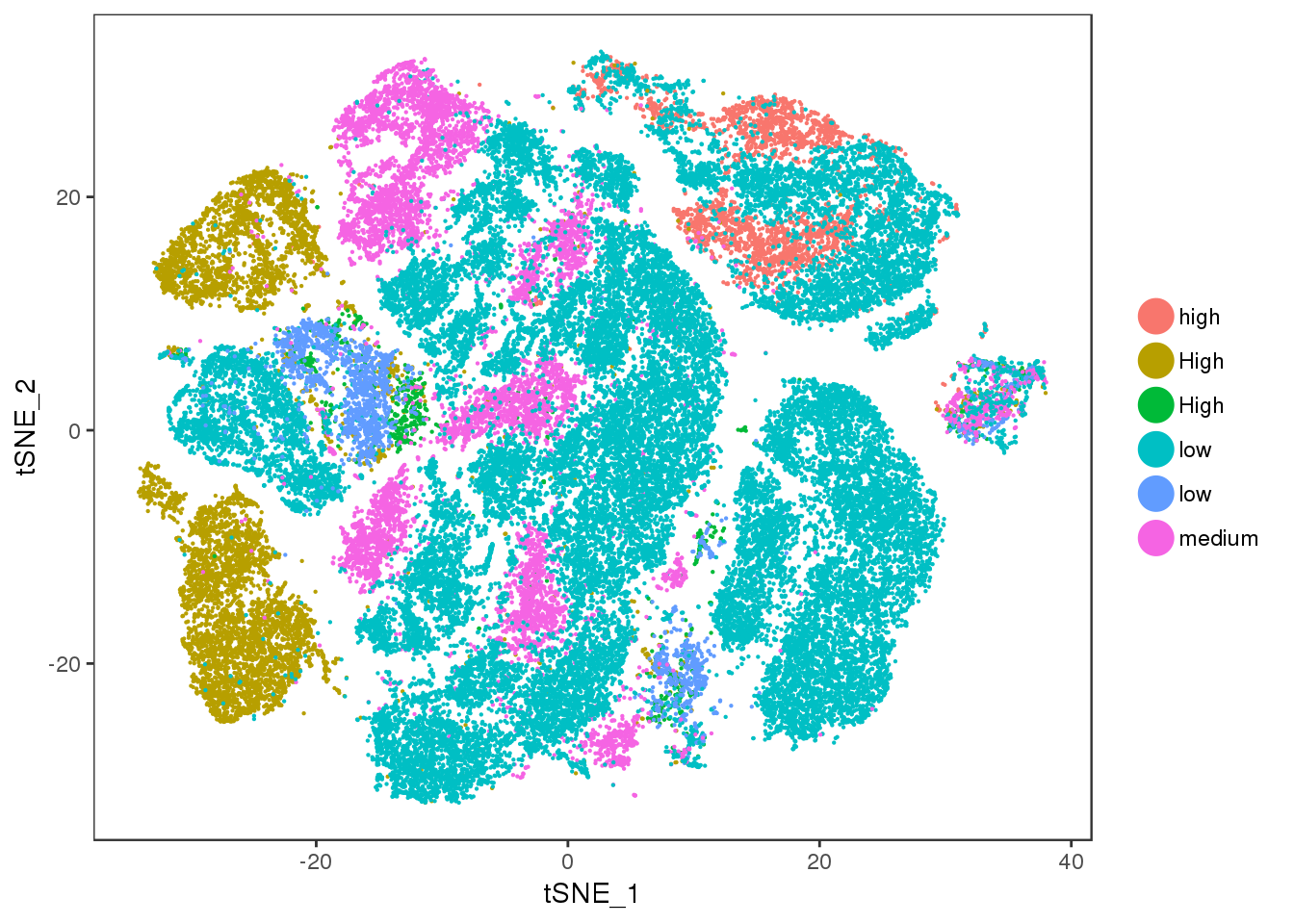
bmi
TSNEPlot(all10x, group.by='bmi', pt.size=0.1)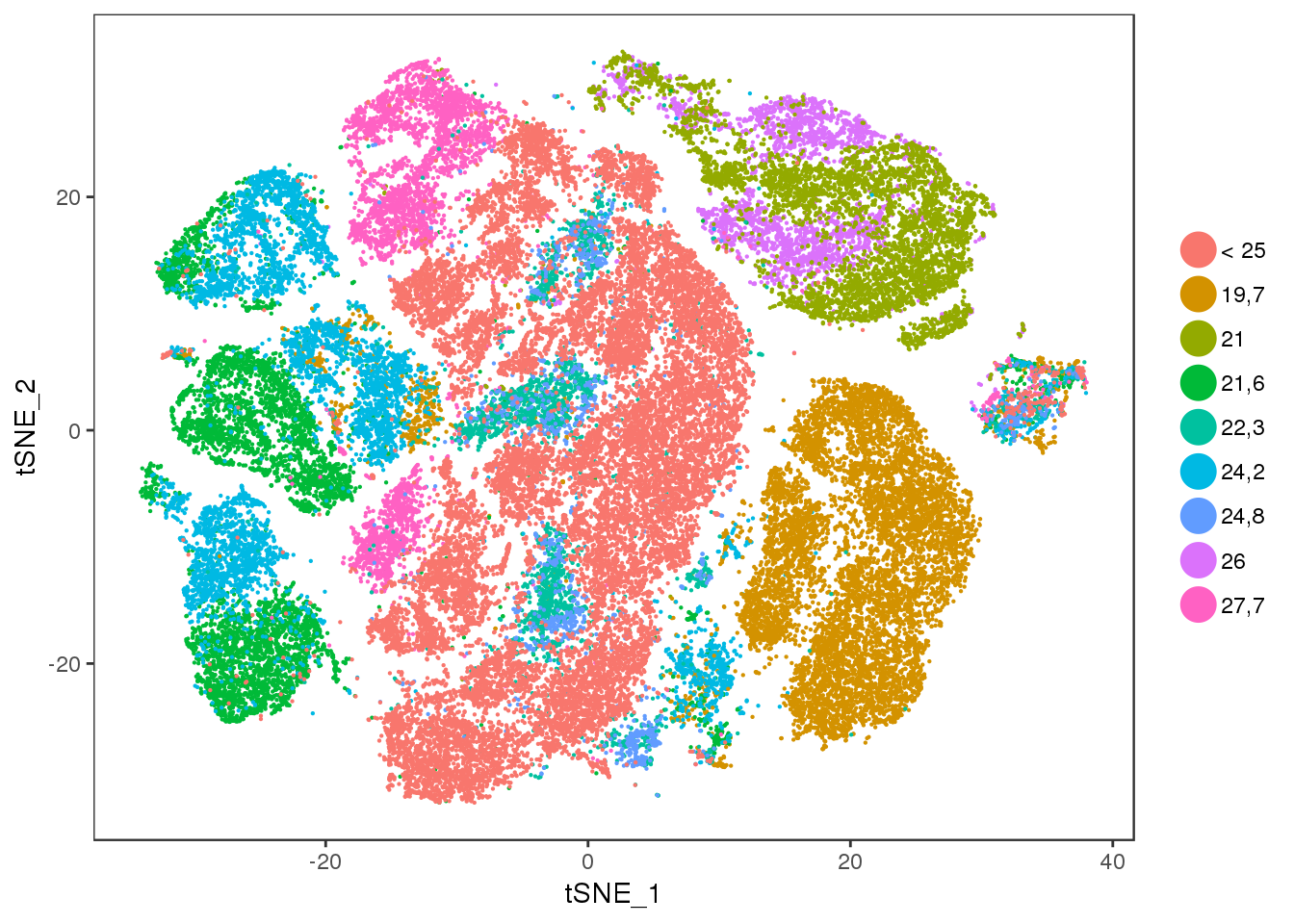
age
TSNEPlot(all10x, group.by='age', pt.size=0.1)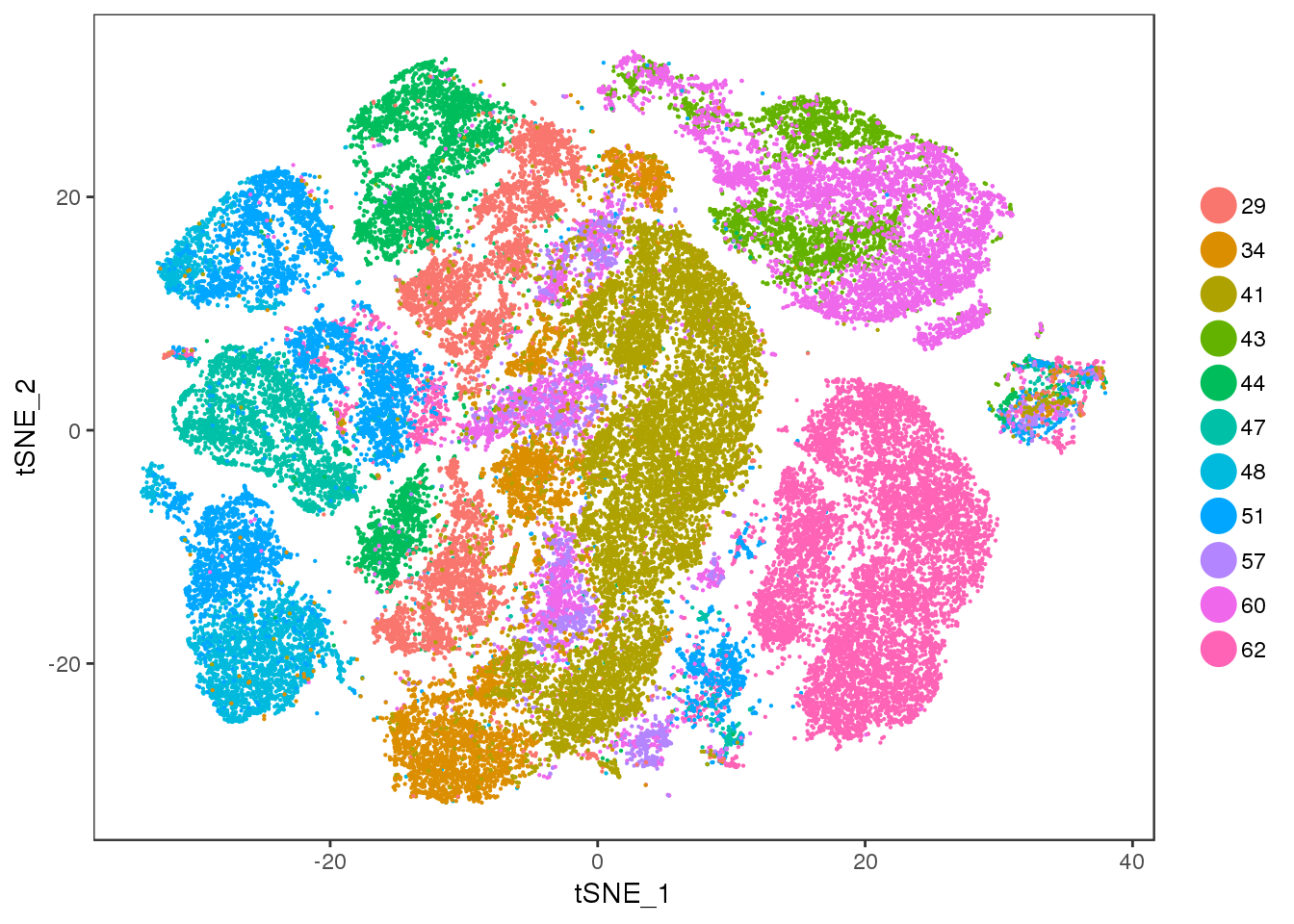
VlnPlot(all10x, group.by='sample_name', features.plot=c('nGene'), point.size.use = -1, x.lab.rot=T)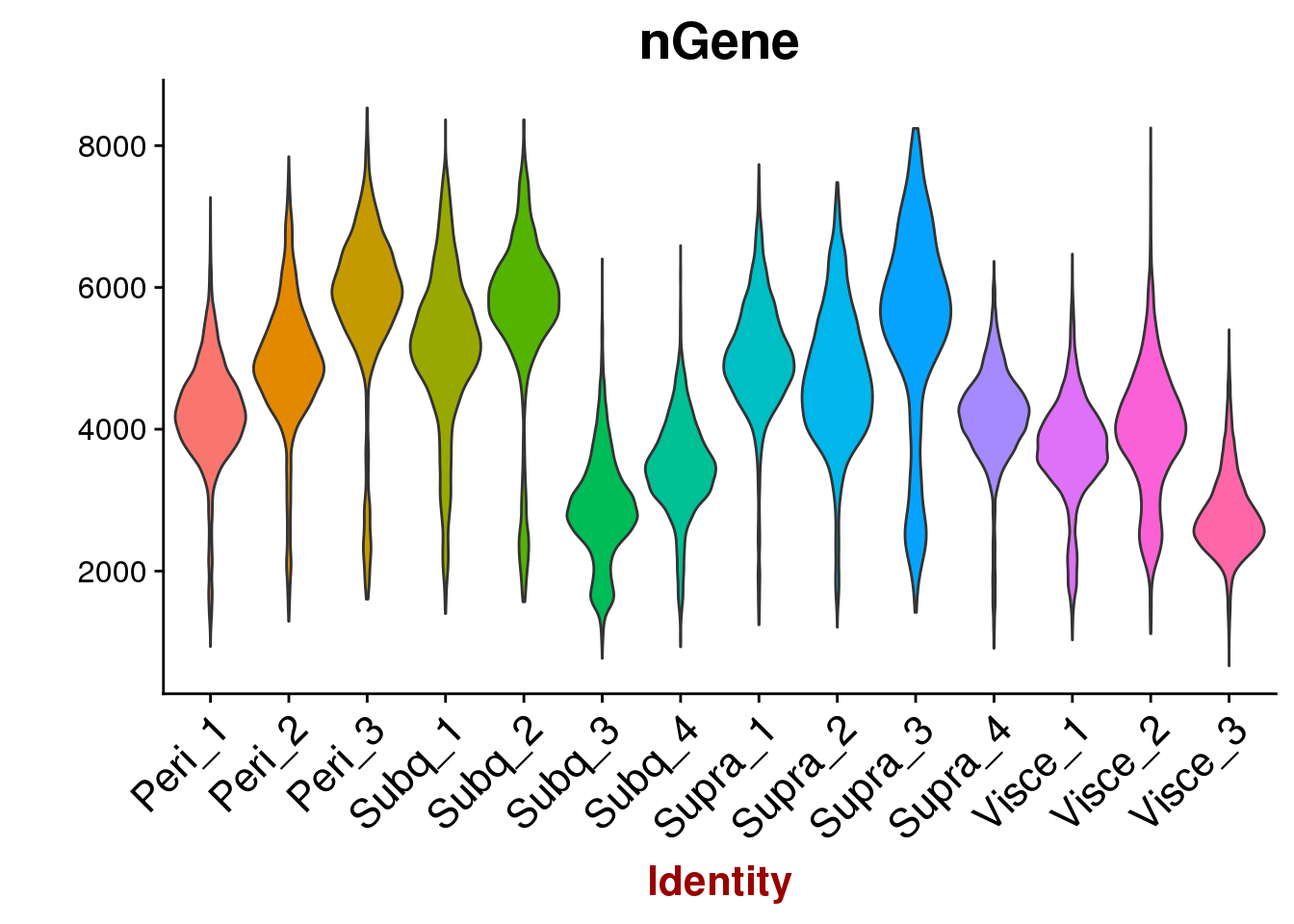
VlnPlot(all10x, group.by='sample_name', features.plot=c('nUMI'), point.size.use = -1, x.lab.rot=T)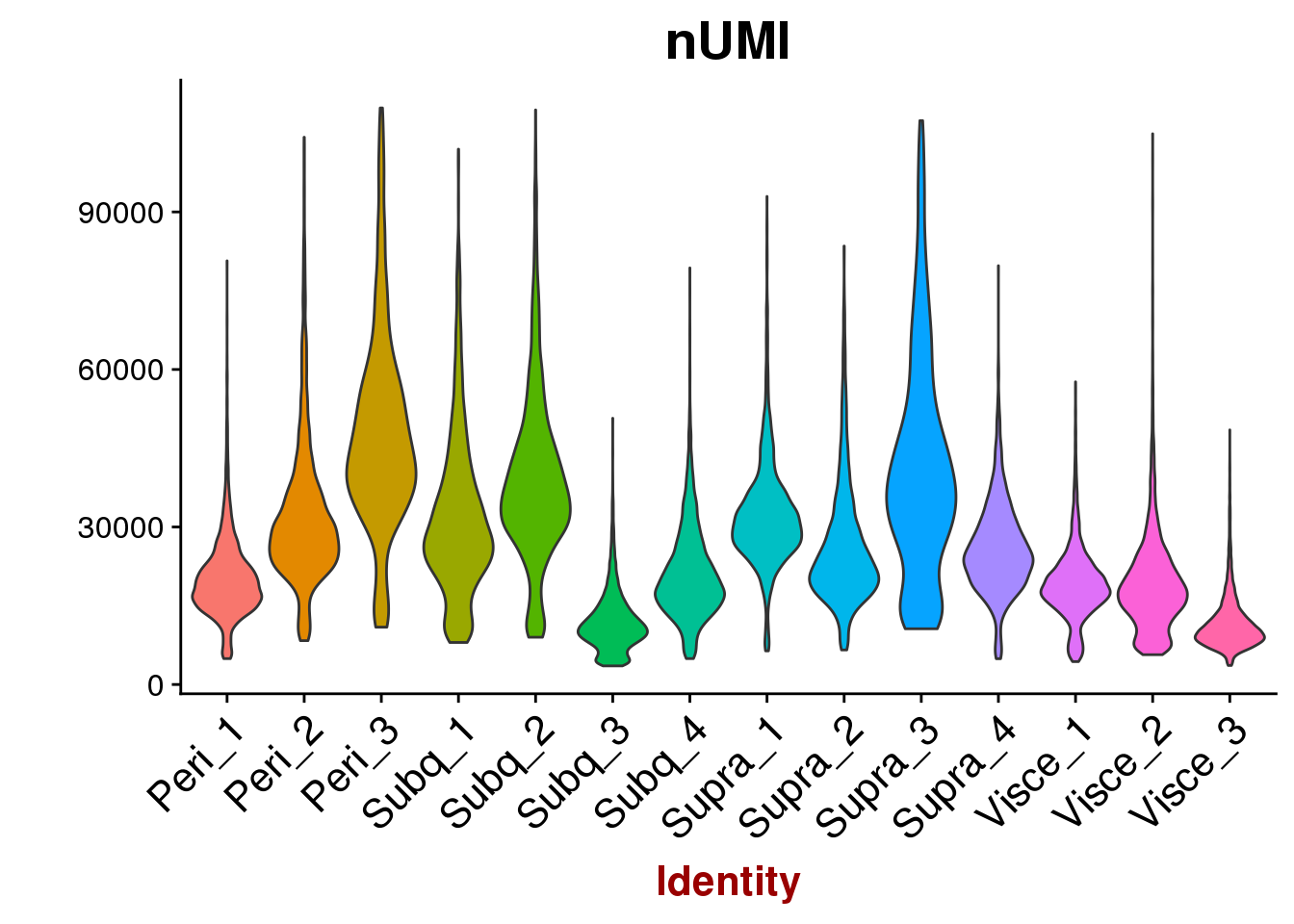
VlnPlot(all10x, group.by='sample_name', features.plot=c('percent.mito'), point.size.use = -1, x.lab.rot=T)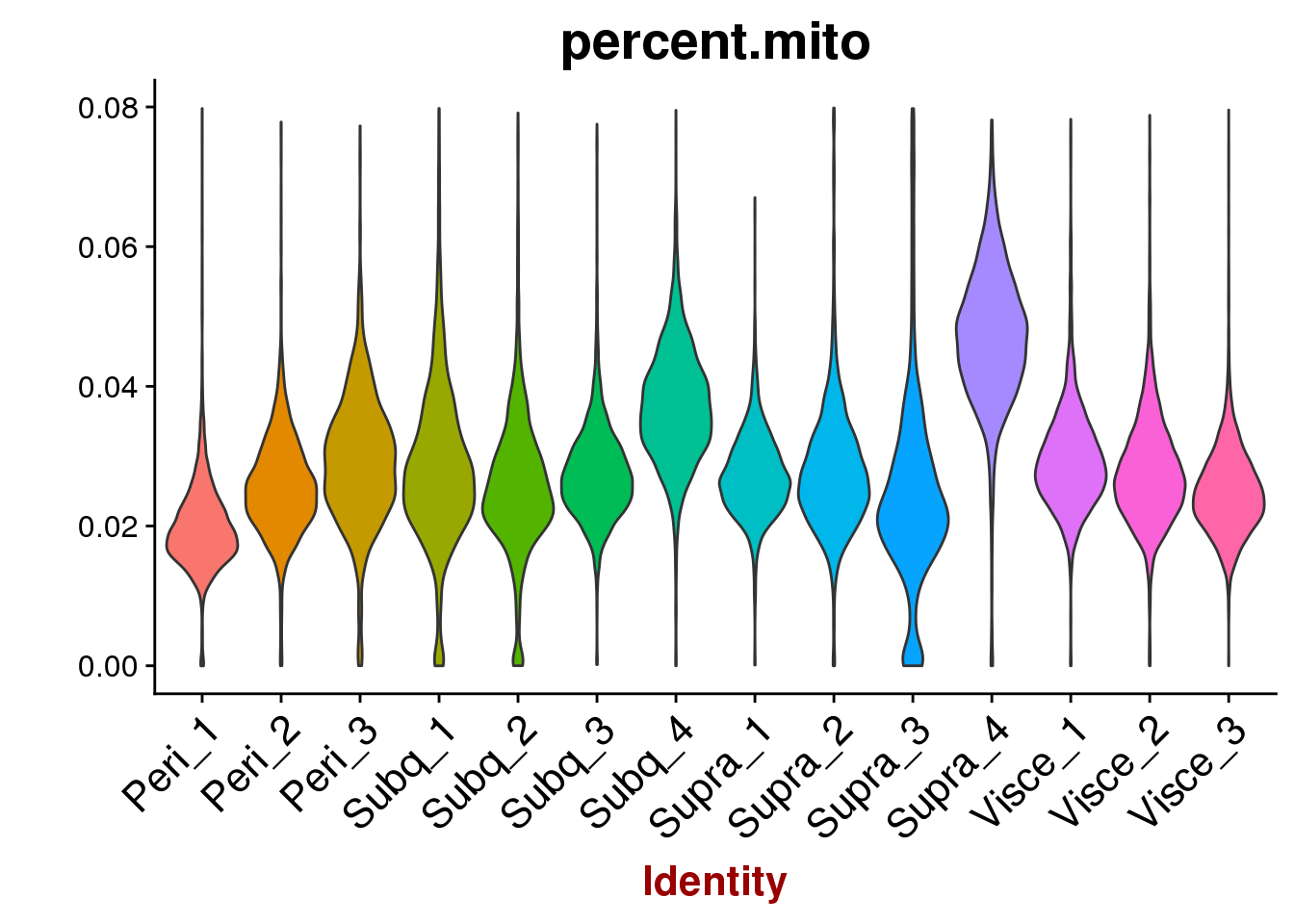
Mixture cluster 12
Sample composition in cluster 12.
cluster12 <- SubsetData(all10x, cells.use=rownames(all10x@meta.data)[which(all10x@meta.data$res.0.5 %in% 12)])
rotate_x <- function(data, column_to_plot, labels_vec, rot_angle) {
plt <- barplot(data[[column_to_plot]], col='steelblue', xaxt="n")
text(plt, par("usr")[3], labels = labels_vec, srt = rot_angle, adj = c(1.1,1.1), xpd = TRUE, cex=1)
}
rotate_x((cluster12@meta.data %>% count(sample_name))[,2], 'n', as.vector(unlist((cluster12@meta.data %>% count(sample_name))[,1])), 45)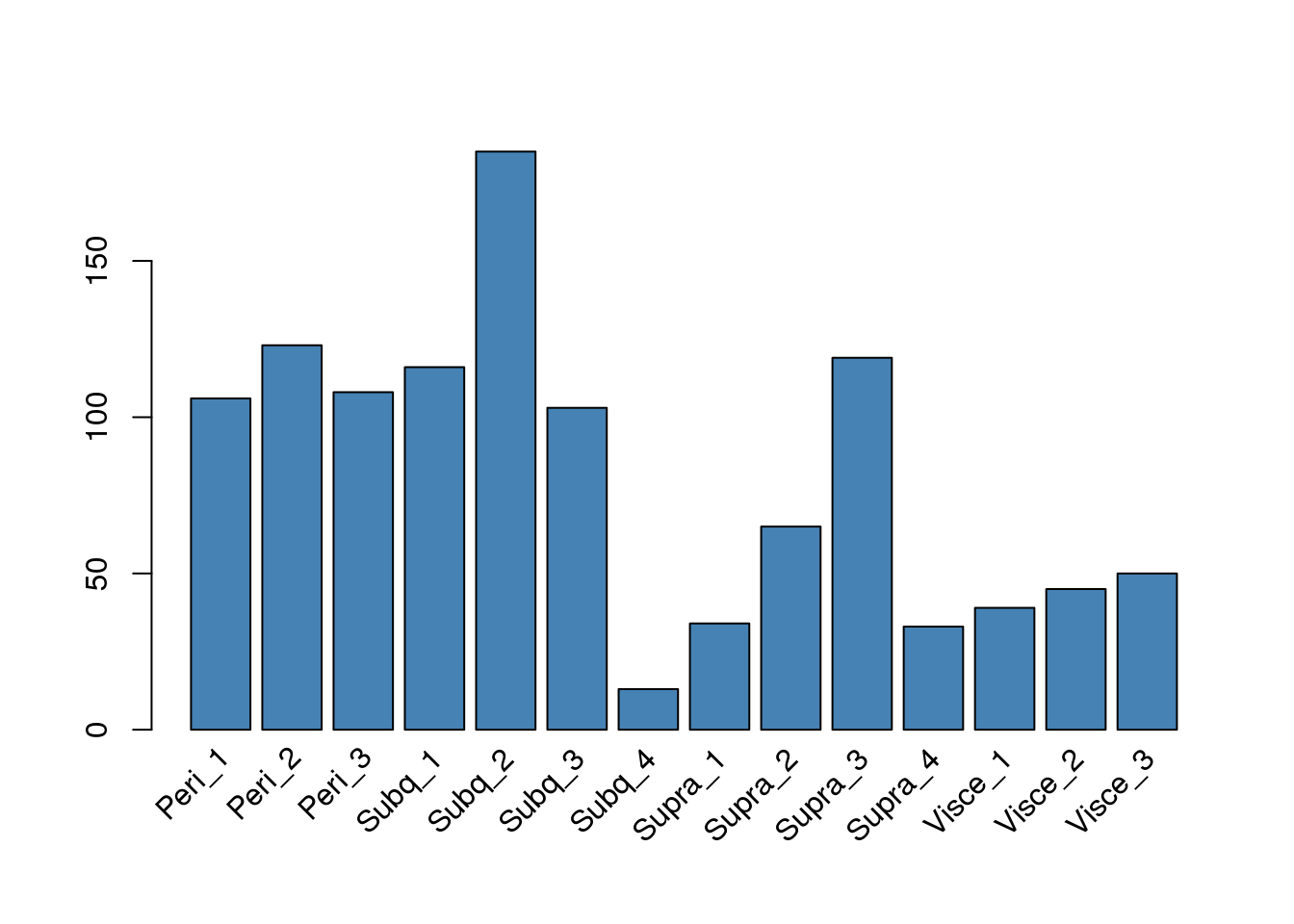
Figures for report
fig1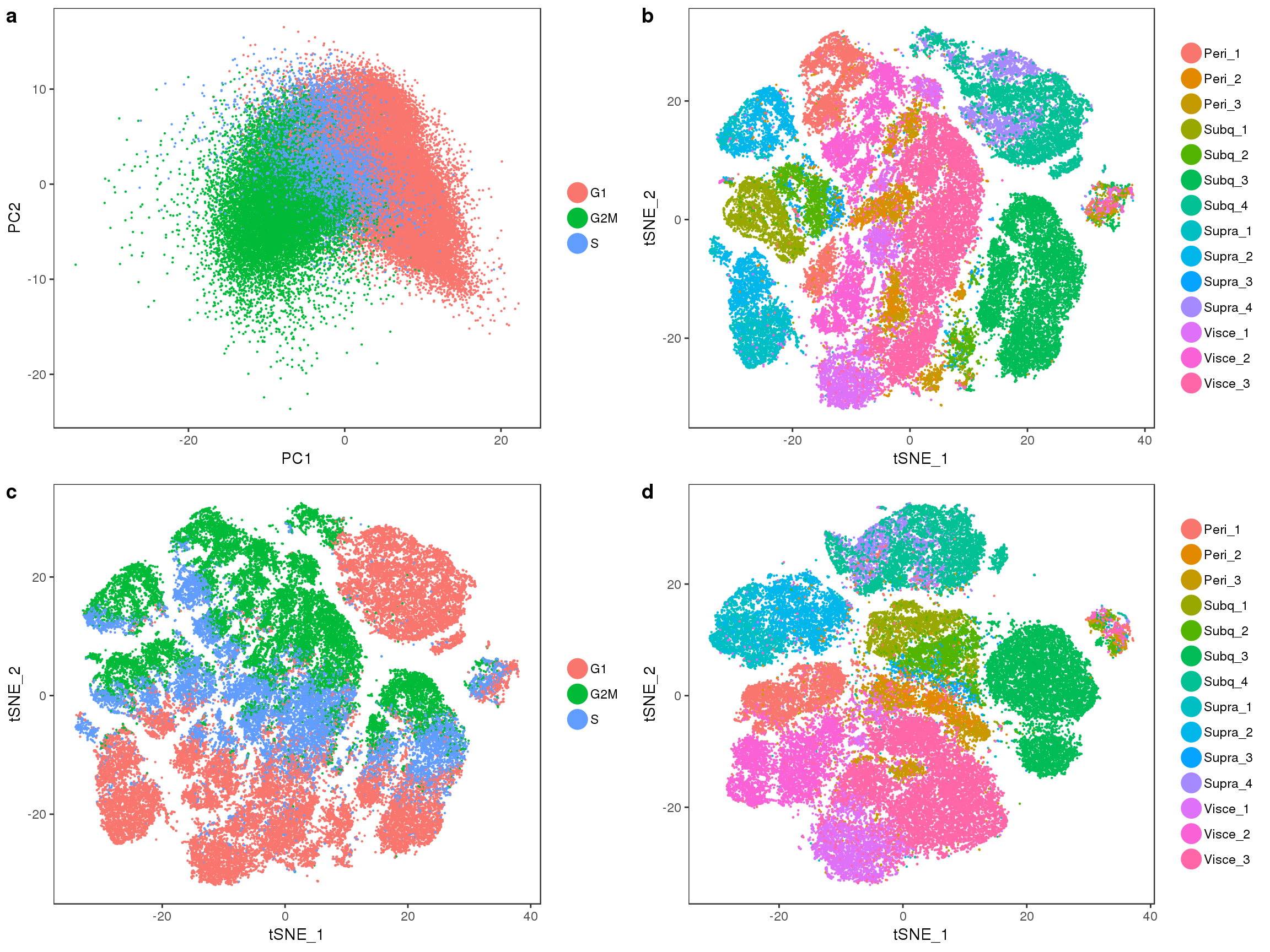
#Supplementary figures
sfig1 <- plot_grid(
VlnPlot(all10x, group.by='sample_name', features.plot=c('nGene'), point.size.use = -1, x.lab.rot=T, size.x.use=8),
VlnPlot(all10x, group.by='sample_name', features.plot=c('nUMI'), point.size.use = -1, x.lab.rot=T, size.x.use=8),
VlnPlot(all10x, group.by='sample_name', features.plot=c('percent.mito'), point.size.use = -1, x.lab.rot=T, size.x.use=8),
labels=c('a', 'b', 'c'), nrow=1
)
save_plot("plots/sfig1_180504_qcplots.pdf", sfig1, base_width=12, base_height=3)
sfig1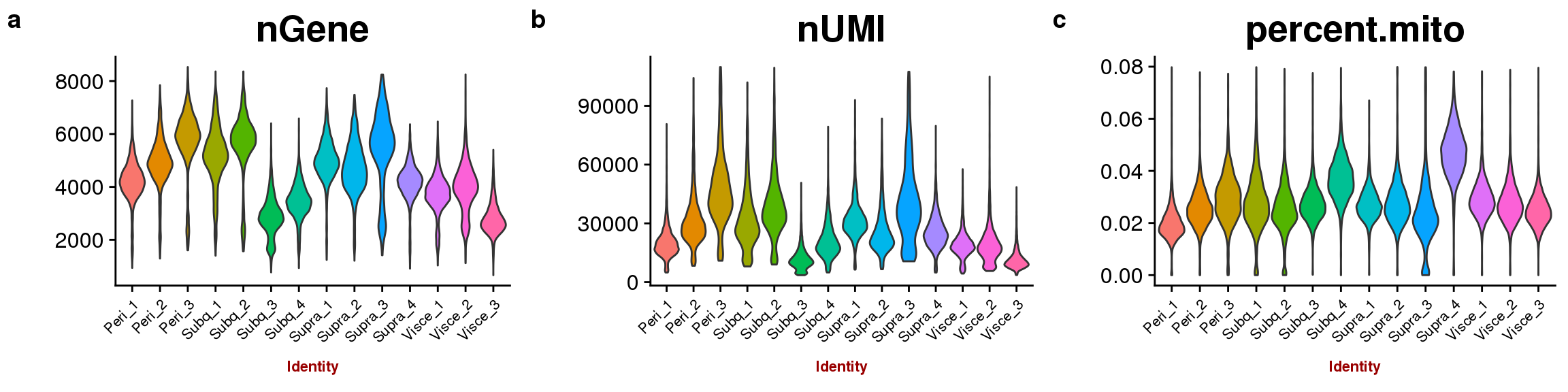
Session information
sessionInfo()R version 3.4.3 (2017-11-30)
Platform: x86_64-redhat-linux-gnu (64-bit)
Running under: Red Hat Enterprise Linux
Matrix products: default
BLAS/LAPACK: /usr/lib64/R/lib/libRblas.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] bindrcpp_0.2.2 dplyr_0.7.6 Seurat_2.3.4 Matrix_1.2-14
[5] cowplot_0.9.3 ggplot2_3.0.0
loaded via a namespace (and not attached):
[1] Rtsne_0.13 colorspace_1.3-2 class_7.3-14
[4] modeltools_0.2-22 ggridges_0.5.0 mclust_5.4.1
[7] rprojroot_1.3-2 htmlTable_1.12 base64enc_0.1-3
[10] rstudioapi_0.7 proxy_0.4-22 flexmix_2.3-14
[13] bit64_0.9-7 mvtnorm_1.0-8 codetools_0.2-15
[16] splines_3.4.3 R.methodsS3_1.7.1 robustbase_0.93-2
[19] knitr_1.20 Formula_1.2-3 jsonlite_1.5
[22] workflowr_1.1.1 ica_1.0-2 cluster_2.0.7-1
[25] kernlab_0.9-27 png_0.1-7 R.oo_1.22.0
[28] compiler_3.4.3 httr_1.3.1 backports_1.1.2
[31] assertthat_0.2.0 lazyeval_0.2.1 lars_1.2
[34] acepack_1.4.1 htmltools_0.3.6 tools_3.4.3
[37] igraph_1.2.2 gtable_0.2.0 glue_1.3.0
[40] RANN_2.6 reshape2_1.4.3 Rcpp_0.12.18
[43] trimcluster_0.1-2.1 gdata_2.18.0 ape_5.1
[46] nlme_3.1-137 iterators_1.0.10 fpc_2.1-11.1
[49] gbRd_0.4-11 lmtest_0.9-36 stringr_1.3.1
[52] irlba_2.3.2 gtools_3.8.1 DEoptimR_1.0-8
[55] MASS_7.3-50 zoo_1.8-3 scales_1.0.0
[58] doSNOW_1.0.16 parallel_3.4.3 RColorBrewer_1.1-2
[61] yaml_2.2.0 reticulate_1.10 pbapply_1.3-4
[64] gridExtra_2.3 rpart_4.1-13 segmented_0.5-3.0
[67] latticeExtra_0.6-28 stringi_1.2.4 foreach_1.4.4
[70] checkmate_1.8.5 caTools_1.17.1.1 bibtex_0.4.2
[73] Rdpack_0.9-0 SDMTools_1.1-221 rlang_0.2.2
[76] pkgconfig_2.0.2 dtw_1.20-1 prabclus_2.2-6
[79] bitops_1.0-6 evaluate_0.11 lattice_0.20-35
[82] ROCR_1.0-7 purrr_0.2.5 bindr_0.1.1
[85] labeling_0.3 htmlwidgets_1.2 bit_1.1-14
[88] tidyselect_0.2.4 plyr_1.8.4 magrittr_1.5
[91] R6_2.2.2 snow_0.4-2 gplots_3.0.1
[94] Hmisc_4.1-1 pillar_1.3.0 whisker_0.3-2
[97] foreign_0.8-70 withr_2.1.2 fitdistrplus_1.0-9
[100] mixtools_1.1.0 survival_2.42-6 nnet_7.3-12
[103] tsne_0.1-3 tibble_1.4.2 crayon_1.3.4
[106] hdf5r_1.0.0 KernSmooth_2.23-15 rmarkdown_1.10
[109] grid_3.4.3 data.table_1.11.4 git2r_0.23.0
[112] metap_1.0 digest_0.6.15 diptest_0.75-7
[115] tidyr_0.8.1 R.utils_2.7.0 stats4_3.4.3
[118] munsell_0.5.0 This reproducible R Markdown analysis was created with workflowr 1.1.1